转cry1C基因抗虫水稻吉生粳3号外源基因整合分析与品系特异性检测
2019-05-17金永梅马瑞于志晶林秀峰
金永梅 马瑞 于志晶 林秀峰
(吉林省农业科学院农业生物技术研究所,长春 130124)
自1996年以来,全球转基因农作物商业化种植面积和销售收入均以倍数增长。水稻是我国乃至全世界最重要的粮食作物之一,目前,全球转基因水稻研究进展迅速,我国转基因水稻研发与世界同步,成功培育出抗螟虫、抗除草剂、耐盐、氮磷高效转基因和优质等系列转基因水稻品种/品系,为我国转基因水稻的产业化提供了技术和品种储备[1]。
通过转基因技术可以将外源基因导入到受体中,改良目标农艺性状。外源基因的插入改变了一个基因与其邻近基因或其邻近染色质的位置关系,随着外源基因的整合区域的不同可发生位置效应,从而影响宿主植物的特定功能及表型。因此,研究T-DNA插入位点的旁侧序列及其插入位点就显得尤为重要[2]。利用农杆菌遗传转化方法导入的外源基因在宿主基因组的插入位点是随机的,每个转化事件中外源基因和基因组DNA拼接组成的旁侧序列具有唯一性。因此,T-DNA旁侧序列分析一直是转基因植物研究的热点,它对于阐述T-DNA整合方式、染色体定位、转基因表达活性等具有重要意义。T-DNA整合过程是个复杂的异常重组过程,包括植物基因组整合位点的删除和重复,T-DNA序列的删除和重复,以及染色体上的易位和到位等现象[3-4]。
染色体步移(Chromosome walking)是指从生物基因组或基因组文库中的已知序列出发,逐步探知其旁邻的未知序列或与已知序列呈线性关系的目标序列的方法。分离旁侧序列的染色体步移方法主要采用基于PCR扩增的染色体步移技术中的半随机引物PCR策略。半随机引物PCR策略中的热不对称交错PCR(Tail-PCR)是将目标序列旁的已知序列中设计的3个嵌套的特定引物(Special primer SP1,SP2,SP3)分别与1个具有低Tm值的随机简并引物(Arbitary degenerate Primer,AD)相结合,根据引物的长短和特异性的差异设计不对称的温度循环,通过分级反应来扩增特异产物的方法,该方法准确性高、操作相对简单[5]。
根据所分离的旁侧序列,通过与NCBI的比对分析确定T-DNA在基因组中的整合位点。该序列是不同转基因作物品系的特异性身份标识,根据其建立的品系特异性检测方法能快速、准确地鉴定不同转基因作物品系[6-7],保证了转基因作物检测的准确性和专一性,为监管转基因作物的育种、生产、加工和销售提供了可靠的技术支撑[8-9]。利用外源基因在受体基因组上整合位点的唯一性,可以建立品系特异性检测方法,该方法可根据外源DNA与旁侧植物基因组连接区序列为靶标设计引物,利用PCR方法扩增出对照和转基因品系特异的PCR条带[10]。
Bt抗虫基因cry1C所编码的杀虫蛋白可以抑制和杀死鳞翅目害虫,是在野生型Cry1Ca5基因的基
础上通过序列优化人工合成的抗虫基因[11]。目前,利用生物技术手段将cry1C基因导入到各种植物中,包括水稻[12-14]、大豆[15-16]、玉米[17-18]等作物,成功获得了对抗鳞翅目害虫具有抗性的转基因植物。前期研究中,我们利用农杆菌介导的遗传转化方法,将cry1C基因导入到水稻品种吉粳88中,获得了抗虫转基因水稻吉生粳3号[19]。本研究旨在通过染色体步移方法获得吉生粳3号的左、右边界旁侧序列,依据旁侧序列确定T-DNA在基因组中的插入位点,并建立吉生粳3号品系特异性PCR方法,为吉生粳3号的身份识别提供理论依据。
1 材料与方法
1.1 材料
转基因水稻吉生粳3号由本课题组培育保存;非转基因水稻吉粳88由吉林省农业科学院水稻研究所提供。
1.2 方法
1.2.1 基因组DNA的提取 将种子播种在1/2MS培养基中,7 d后取样保存。采用CTAB方法提取0.1g水稻样品中的基因组DNA[20]。DNA样品的纯度和浓度用NanoDrop进行检测。
1.2.2 转基因植株吉生粳3号的Southern Blot 利用CTAB法提取水稻叶片的基因组DNA,利用DIGHigh Prime DNA Labeling and Detection Starter Kit I(Roche公司)进行Southern杂交,方法参考试剂盒说明书。具体步骤为:取60 μg基因组DNA,用限制性内切酶SacI(TaKaRa)进行完全酶切。酶切产物用0.8% 琼脂糖凝胶电泳分离后转膜(Amersham),用地高辛标记的cry1C探针进行杂交,再用X-光胶片化学发光自显影。
扩增探针引物序列为:cry1C-F:5'-TTCTACTGGGGAGGACATCG-3',cry1C-R :5'-CGGTATCTTGGGTGATTGG-3'。
1.2.3 转基因水稻吉生粳3号的右边界旁侧序列的获取 采用染色体步移试剂盒(TaKaRa GenomeWalking Kit,Code No.6108)分离吉生粳3号中外源载体插入位点处的旁侧序列。在已知的T-DNA序列右边界设计3条同向且退火温度较高的特异性引物(表 1)。
以1 μL(200 ng 左右)吉生粳3号基因组DNA为模板,使用右边界特异性引物和试剂盒中的简并引物AP1、AP2、AP3、AP4进行巢式PCR。

表1 T-DNA右边界巢式引物序列
巢式PCR扩增体系为:1st PCR 反应在50 μL体系中进行,以1 μL基因组DNA,0.5 μL TaKaRa LA Taq(5 U/μL),5 μL 的 10×LA PCR Buffer II(Mg2+plus),8 μL 的 dNTP Mixture(2.5 mM each),1 μL的 AP1、AP2、AP3、AP4 引 物(100 pmol/μL),1 μL的SP1引物(10 pmol/μL)进行扩增;2nd和3rd PCR反应分别将上一轮PCR反应液稀释1 000倍后,取 1 μL作为 PCR 反 应 模 板,1 μL的 AP1、AP2、AP3、AP4 引物(100 pmol/μL)分别与 1 μL 的 SP2和SP3引物(10 pmol/μL)进行扩增PCR扩增体系同上;巢式PCR反应扩增程序按照染色体步移试剂盒(TaKaRa GenomeWalking Kit,Code No.6108) 中的方法进行。
取1st,2nd,3rd PCR反应液各5 μL,使用1%的琼脂糖凝胶进行电泳,分析PCR扩增条带并筛选有义引物组合,切胶回收被选条带,利用pMDTM 19-T Vector(Takara,Code No.6013)进行 TA 克隆(TaKaRa DNA Ligation Kit,TaKaRa,Code No.6022),阳性质粒进行测序获得右边界旁侧序列,测序引物为5'-CCAGTTCAGCTAATCTTCTT-3'。引物合成及PCR扩增条带的测序均由宝生物有限公司完成。
1.2.4 旁侧序列比对分析及插入位点的确定 利用NCBI BLAST 软件(http://www.ncbi.nlm.nih.gov/BLAST),将PCR扩增条带的测序结果分别与水稻基因组序列和T-DNA序列进行同源比对分析,确定外源T-DNA在吉生粳3号基因组中的插入位点。
1.2.5 吉生粳3号的左边界旁侧序列获取 在吉生粳3号基因组中T-DNA插入位点的左侧设计正向引物(C10-1496F),在T-DNA左边界设计3条同向巢式引物(C10F-SP4,C10F-SP5,C10F-SP6),进行PCR扩增以进一步确认插入位点的正确性。引物序列见表2。
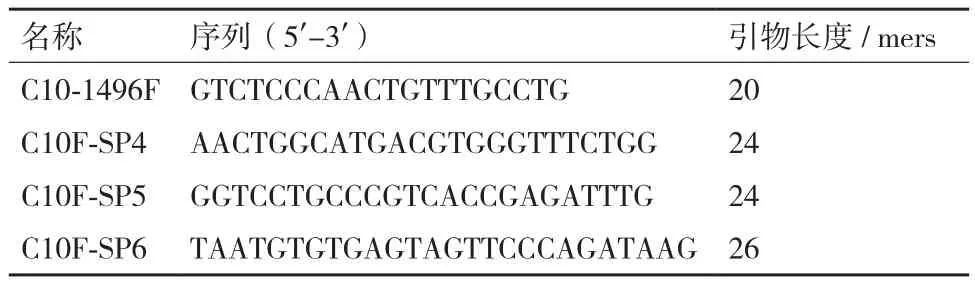
表2 T-DNA左边界巢式引物序列
PCR 扩增体系为:总体系20 μL,康为世纪2×Es Taq MasterMix 10 μL(含有 Es Taq DNA Polymerase,PCR buffer,3 mmol/L MgCl2,400 μmol/L dNTP mix),引物各 1 μL(10 μmol/L),水稻 DNA 2 μL(100 ng/μL),剩余用ddH2O 补足;PCR 扩增程序为,94℃ 1 min;94℃ 10 s,55℃ 1 min,72℃ 40 s,35 个循环 ;72℃ 10 min。PCR产物经过回收、纯化、测序获得左边界旁侧序列。
1.2.6 吉生粳3号特异性PCR检测 提取非转基因对照品种(吉粳88)和吉生粳3号基因组DNA进行特异性PCR检测。PCR反应在25 μL体系中进行 :12.5 μL 的 康 为 世 纪 2×Es Taq MasterMix( 含有 Es Taq DNA Polymerase,PCR buffer,3 mmol/L MgCl2,400 μmol/L dNTP mix),1 μL 10 μmol/L 正向引物,1 μL 10 μmol/L 反向引物,1 μL T-DNA 特异性引物,水稻基因组 DNA 2 μL(100 ng/μL),剩余用ddH2O 补足。反应条件为:94℃ PCR预变性2 min,94℃变性 30 s,55℃退火 30 s,35循环,72℃10 min,引物序列如下:正向引物为C10-F:TGCACGCCTACTCCACATTA,反向引物为C10-R:TGCTTGCTAGACTCACCACA,T-DNA特异性引物为C10-TDNAR:TCACCACTCGATACAGGCAG。
2 结果
2.1 转基因水稻吉生粳3号右边界旁侧序列及插入位点
通过农杆菌遗传转化法,将Bt抗虫基因cry1C的P3300-cry1C植物表达载体,导入到吉林省水稻品种吉粳88中,获得了抗虫转基因水稻独立转化植株。通过分子检测和抗性分离比分析,从49个独立转化植株中筛选出8个单拷贝株系。分别提取和非转基因水稻和吉生粳3号的基因组DNA,用限制性内切酶SacI和DraI分别进行酶切,地高辛标记的cry1C探针进行杂交,筛选出外源目的基因cry1C为单拷贝的转基因水稻株系吉生粳3号(图1)。

图1 植物表达载体示意图与Southern blot
为了获取吉生粳3号右边界旁侧序列,提取基因组DNA,利用染色体步移方法获得PCR特征条带。T-DNA序列右边界3条同向巢式引物C10R-SP1、C10R-SP2、C10R-SP3,分别与简并引物AP1、AP2、AP3、AP4进行3轮PCR扩增。结果(图2)表明,3轮PCR所有引物组合均获得比较清晰的PCR条带,但只有简并引物AP2与3条巢式引物组合所获得的条带大小呈递减趋势(泳道4-6),因此可以判断为吉生粳3号右边界旁侧序列的特异性条带。
切胶回收C10R-SP3与AP2引物组合扩增出的PCR条带(图2-B中泳道6,约1.8 kb),通过TA克隆方法获得包含目的片段的载体,经过目的片段的测序最终获得目的片段的核苷酸序列。
将吉生粳3号右边界旁侧序列(图3-A),利用NCBI数据库中的 BLAST工具进行比对,确定了T-DNA在吉生粳3号基因组中的插入位点。比对分析结果表明,吉生粳3号右边界旁侧序列中1-67 bp区段与载体序列右边界的部分序列有100%的相似性,68-732 bp区段与水稻基因组(日本晴)2号染色体上的基因间区(非编码区)2 790 589-2 789 925 bp区段具有99%的相似性,因此插入位点初步可以判断为2 790 589(图3-B)。
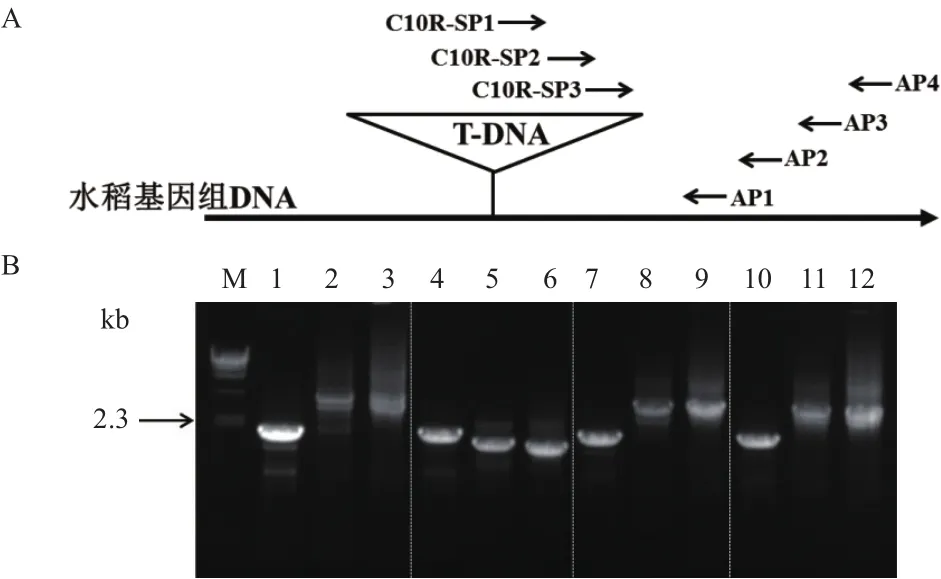
图2 吉生粳3号右边界旁侧序列巢式PCR
2.2 吉生粳3号左边界旁侧序列及插入位点
根据吉生粳3号右边界旁侧序列确定的插入位点,在吉生粳3号插入位点左侧基因组中设计正向引物(C10-Chr2-1496F),在T-DNA左边界区段设计3条同向T-DNA特异性巢式引物(C10F-SP4,C10F-SP5,C10F-SP6)(表 2)进行 PCR 扩增,进一步确认插入位点的正确性。PCR结果获得了3条大小递减的PCR特征条带,对其中一条PCR条带(泳道3)进行了回收、克隆、测序(图4)。测序获得的序列分别与载体序列和水稻基因组序进行列比对确定插入位点,结果插入位点确定为水稻基因组(日本晴)2号染色体上的2790685。
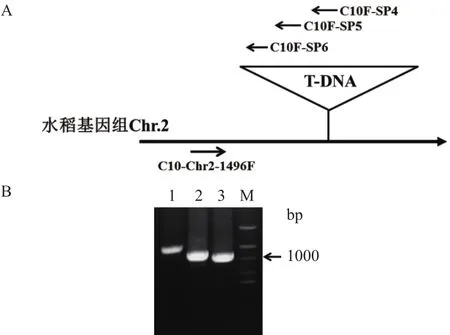
图4 吉生粳3号左边界PCR
2.3 吉生粳3号的T-DNA插入位点及品系特异性PCR检测
根据右边界旁侧序列可以判断T-DNA在吉生粳3号基因组中的插入位点为2号染色体上的2 790 589位,根据左边界旁侧序列可以插入位点为2号染色体上的2 790 685位。因此,根据左、右边界旁侧序列可以确定T-DNA的插入位点为2号染色体上的2 790 685-2 790 589位点,水稻基因组序列发生了96个碱基的缺失(图5-A)。
在吉生粳3号基因组中T-DNA的插入位点两侧分别设计正向引物和反向引物(C10-F和C10-R),在T-DNA的左边界设计特异性引物(C10F-TDNAR),进行吉生粳3号特异性PCR检测。提取非转基因对照、吉生粳3号、和其它两个转基因水稻株系的基因组DNA,用上述3条引物同时进行PCR扩增。在PCR扩增反应程序中延伸时间限定为40 s,因此在吉生粳3号中引物C10-F和C10F-TDNAR可进行扩增,在非转基因对照和其它基因转基因水稻株系中引物C10-F和C10-R可进行扩增。PCR结果(图5-B)发现,转基因水稻吉生粳3号中扩增出一条428 bp的转基因特异性片段,非转基因对照和其它两个转基因株系中扩增出一条1 023 bp的非转基因特异性片段,与预期大小一致,说明引物C10-F、C10-R、C10F-TDNAR能够特异性地识别吉生粳3号水稻。

图5 T-DNA在吉生粳3号基因组中的插入位点及品系特异性PCR
3 讨论
在转基因生物中外源基因的T-DNA旁侧序列具有非常重要的意义,利用它可以分析T-DNA在宿主生物基因组中的整合方式、染色体定位、对内源基因的表达影响等情况。目前,分离旁侧序列的方法有Tail-PCR、反向PCR、质粒拯救、染色体步移、全基因组测序等。李亚丽等[21]通过Tail-PCR方法分离了转mCherry基因水稻T-DNA旁侧序列并确定了它在基因组中的整合位点;Cao等[22]利用染色体步移方法分离转基因小麦品系B10201-2的旁侧序列,并建立了品系特异性PCR方法;Guo等[23]利用全基因组测序方法,在转基因大豆中分离了G2EPSPS基因和GAT基因的旁侧序列并确定了基因组中的插入位点。
转基因植物中外源基因的整合区域与外源基因的稳定表达及宿主基因组中上下游基因的正常表达密切相关。如果T-DNA整合进入内含子,外源基因容易造成基因沉默,导致目的基因不表达;如果T-DNA整合进入功能基因内部,会影响功能基因的正常表达并会对基因功能造成影响。因此,转化事件筛选过程中的众多指标中选择T-DNA整合进入基因间区的转化事件是重要指标之一。有研究表明,外源基因片段易整合进入植物基因组中转录活跃区域或染色体的末端[24-25];在转基因拟南芥中T-DNA整合位点与染色体上基因分布的密度相关,T-DNA插入位点倾向于基因间区域,整合在外显子与内含子区域的频率没有显著性差异[26]。为了分析外源基因T-DNA在抗虫转基因水稻吉生粳3号中的整合区域与插入位点,我们通过染色体步移方法分离了吉生粳3号的右边界旁侧序列。根据右边界旁侧定位外源基因在转基因水稻吉生粳3号基因组中的插入位点并分离出右边界旁侧序列。由于吉生粳3号转基因水稻的外源基因T-DNA区域插入到水稻基因组2号染色体的基因间区,因此,外源片段的插入不会使吉生粳3号产生位置效应。通过对转基因水稻吉生粳3号多个世代的外源目的基因遗传稳定性分析表明,cry1C基因在DNA水平、RNA水平和蛋白水平上表达稳定;农艺性状和目标性状鉴定分析发现,除了抗虫性之外其他农艺性状与对照品种没有显著差异,并且保持稳定,说明T-DNA在吉生粳3号基因组中的插入位点对外源基因自身的稳定表达和水稻內源基因的表达均没有造成不利影响(未发表数据)。
吉生粳3号的左右边界旁侧序列分析表明,在外源基因的整合过程中T-DNA右边界旁侧序列的断裂点在A、T碱基处,左边界断裂点在C、T碱基处,水稻基因组序列缺失了96 bp,载体序列未发生碱基缺失或易位等变化。有研究表明转基因水稻T-DNA左右边界及边界内侧序列的断裂点主要集中在A、T碱基处[20];Tzfira等[27]认为T-DNA 整合进宿主基因组的主要路径为双链断裂修复机制,T-DNA整合过程中农杆菌中的稀有位点限制性内切酶在宿主中表达,使宿主基因组DNA产生双链断裂,并且在宿主体内消化T-DNA,导致宿主基因组序列的缺失和T-DNA的丢失。
转基因作物品系特异性检测方法特异性高,适合转基因产品的定性和定量检测,是对转基因植物进行监督管理、保障其健康发展的重要技术基础。郭超等[28]利用hiTail-PCR方法获得了转基因水稻BarKasalath-01中外源基因在基因组上的旁侧序列并建立了该转化事件特异性检测方法;金芜军[29]等人于2013年基于转基因抗虫水稻品系TT51-1的旁侧序列建立了鉴定纯合型转基因抗虫水稻品系TT51-1 的方法。本研究中,根据外源基因T-DNA在吉生粳3号基因组中的整合位点,在整合位点两侧和T-DNA左边界共设计3条引物,建立了吉生粳3号品系特异性PCR检测方法。本研究所建立的cry1C转基因吉生粳3号品系水稻特异性检测方法,对提高该品系的鉴定效率及准确性具有重要意义。
4 结论
本研究利用染色体步移方法,获得转cry1C基因抗虫水稻吉生粳3号的外源基因旁侧序列及其水稻基因组中的插入位点,并建立了吉生粳3号品系特异性PCR检测方法。该结果为吉生粳3号的身份识别提供了准确、快速的检测技术手段。
