玉米C型细胞质雄性不育花药不同发育时期的转录组分析
2019-05-08薛亚东杨露杨慧丽李冰林亚楠张怀胜郭战勇汤继华
薛亚东,杨露,杨慧丽,李冰,林亚楠,张怀胜,郭战勇,汤继华,2
玉米C型细胞质雄性不育花药不同发育时期的转录组分析
薛亚东1,杨露1,杨慧丽1,李冰1,林亚楠1,张怀胜1,郭战勇1,汤继华1,2
(1河南农业大学农学院/省部共建小麦玉米作物学国家重点实验室,郑州 450002;2长江大学主要粮食作物产业化湖北省协同创新中心, 湖北荆州 434025)
【目的】通过分析玉米C型胞质雄性不育“三系”材料花药不同发育时期的转录组数据,以期阐明玉米C型胞质的不育和恢复机制,并解析不育基因与恢复基因之间相互作用的调控网络,为玉米C型细胞质雄性不育在不育化制种中的利用提供理论依据。【方法】以中国玉米生产上的骨干自交系豫自87-1为背景的C型胞质不育系、保持系、恢复系为材料,通过对3种材料减数分裂的前期Ⅰ、中期Ⅰ及末期Ⅱ(四分体)时期的花药进行转录组测序并利用hisat2、ballgown及DESeq2等工具进行生物信息学分析,寻找三系花药不同时期、相同时期不同材料间以及发育时序中差异表达的基因,预测C型胞质不育机制与育性恢复的调控网络;同时通过实时定量PCR对测序分析结果进行验证;通过酶活测定验证推测的C型胞质不育及恢复假说。【结果】所有材料的转录组测序共产生156.59 Gb的序列数据,比对并组装共得到53 035个基因;在恢复系与不育系、保持系与不育系以及“三系”花药不同时期之间共筛选出非重复差异基因5 676个,其中发育阶段差异基因4 705个,同时期材料间差异基因2 693个,发育时序差异基因135个。GO分子功能分析显示ATP和DNA结合相关的基因和锌离子结合基因得到高度富集;细胞组分中膜基本组分、核内及质膜内的基因得到富集;以DNA为模板的转录、转录调控、氧化还原及初级代谢等生物学过程中的基因得到富集。KEGG分析表明,差异基因主要富集于氧化磷酸化、碳代谢及糖酵解等能量代谢相关的途径中。不育系相对保持系而言,多个与氧化磷酸化相关的基因下调表达,而恢复系中不但相应基因的表达水平得到恢复,而且同时协调调节了同一能量代谢途径中的其他基因,定量分析显示差异基因的表达差异及趋势与转录组测序结果基本一致。ATP酶活结果表明不育系相比保持系,ATP酶活显著降低,恢复系中由于恢复基因的作用其活性得到大幅恢复。【结论】玉米C型胞质不育基因引起基因表达变化可能发生在减数分裂中期Ⅰ之后,末期Ⅱ之前;玉米C型胞质不育的形成可能是由于不育基因引起的能量亏损所致,而恢复基因则通过能量补偿促使育性得以恢复。
玉米;C型细胞质雄性不育;转录组;差异表达基因;调控网络
0 引言
【研究意义】玉米是中国第一大粮食作物,常年播种面积333万hm2以上,年用种量7.5亿kg左右,需要制种23万hm2左右(全国农业技术推广服务中心 www.natesc.org.cn)。由于玉米种子生产多数采用人工去雄的方式,人工去雄的直接费用约6亿元,导致种子生产成本增加,在一定程度上制约着中国种子企业的国际竞争力,而不育化制种是当前及今后种子企业提高竞争力的一个重要途径。在玉米的质核互作不育类型中,C型细胞质雄性不育因其具有不育性稳定、恢复彻底等特点,得到了玉米育种家及相关种子企业的高度关注。由于玉米C型胞质雄性不育及恢复机制目前仍不清楚,从而在一定程度上限制了其在种子生产上的大面积推广应用。因此,明确玉米C型细胞质不育和恢复的分子机制及其调控网络,将对玉米C型胞质雄性不育的应用具有一定的促进作用。【前人研究进展】植物尤其是作物中细胞质雄性不育材料是杂交种子生产的一种重要种质资源,同时也是研究核质互作的重要遗传材料,因而得到了遗传学家和育种家的广泛关注。前人曾经对细胞质雄性不育开展了大量的研究,玉米T型细胞质的不育基因是第一个被鉴定的细胞质雄性不育基因[1]。水稻先后克隆了CMS-HL不育基因[2]、CMS-BT的不育基因[3]以及CMS-WA的不育基因[4]。油菜中应用较为广泛的CMS-pol、CMS-nap与CMS-ogu 3种类型的细胞质雄性不育的不育基因、和已经克隆[5-7]。萝卜、油菜、小麦等其他作物的细胞质雄性不育相关的部分基因也已被鉴定。目前,已经克隆的植物不育基因均与线粒体基因相关,且均为重组形成的嵌合基因。与之相应的恢复基因除玉米的[8]、水稻[9]和[10]、甜菜的[11]外,均为编码PPR基序蛋白的基因[3,12-17]。根据已有的研究可将细胞质雄性不育机理归纳为3个假说:毒性假说、能量亏缺假说及细胞异常凋亡假说;其相应的恢复基因则通过转录后水平的编辑、剪接、多聚腺苷酸化、剪切等方式,或翻译及翻译后水平的修饰来补偿或逆转不育基因的危害,进而恢复植株的育性[18]。玉米C型细胞质雄性不育有2个重叠效应的恢复基因和[19],分别定位于第8染色体短臂和第5染色体长臂上[20]。玉米C型胞质线粒体基因组中具有多个区域重复并伴有重组,造成与线粒体呼吸链复合体相关基因具有多个拷贝,如、或形成嵌合基因,如,推测其中可能包含不育基因[21-22]。光学及电子显微镜观察结果表明,C型胞质不育系小孢子的败育发生在单核早期[23-24],而在二分体时期绒毡层细胞出现U氏小体异常[24],及至四分体绒毡层出现大量不规则液泡[23-24]。全基因组转录分析能够鉴定到与育性恢复及不育相关的功能基因及其调控与代谢的网络。已报道的雄性不育转录组分析结果表明,洋葱细胞质雄性不育与线粒体氧化磷酸化有关[25],辣椒细胞质雄性不育主要涉及ATP合酶、NADH脱氢酶及细胞色素氧化酶[26]。对玉米C型胞质不育系与保持系的花粉母细胞时期及单核期花药转录组分析发现MYB转录因子、氨基酸代谢和脂肪酸合成途径可能对育性产生重要影响[27]。【本研究切入点】尽管前人确立了C型胞质花粉败育时期以及分析了花粉母细胞时期与单核时期在不育系与保持系之间的差异基因,但C型胞质雄性不育的分子机制尚未解析;由于未能同时对同基因型的恢复系进行分析,无法全面解析不育与恢复可能的代谢和调控机制;而对于孢子体不育材料,在花粉败育时期取样,无法分析败育形成的基因。【拟解决的关键问题】本研究拟通过细化取样时期,比较玉米C型胞质三系材料在减数分裂前期Ⅰ、中期Ⅰ及四分体时期花药的转录组数据,在转录水平上分析玉米C型胞质雄性不育及恢复的机制,明确玉米C型胞质雄性不育发育过程中基因表达规律,以期为玉米C型雄性不育及恢复分子机制研究提供依据,同时为恢复基因及不育基因的克隆及功能分析奠定基础。
1 材料与方法
1.1 试验材料
选用以优良玉米自交系豫自87-1为背景构建的玉米细胞质雄性不育系CMS-Es87-1()、保持系N87-1()及恢复系CMS-Es87-1()为试验材料。试验材料于2016年春分期种植于河南农业大学科教园区。播种40 d后,参考Ma等[28]方法每隔2 d检查玉米雄穗的花药发育进程,待花药发育至减数分裂时期将玉米整株取回实验室。对雄穗局部区域进行三点(区域两头及中部)醋酸洋红染色镜检,选取减数分裂前期(P1)、减数分裂中期Ⅰ(M1)及四分体时期(T2)的花药,液氮速冻后-80℃保存备用。每一个材料每一个时期各取3个生物学重复进行转录组测序,每个生物学材料由多个植株的花药组成,以消除个体及环境的差异。
1.2 花药RNA提取
采用TRIzol(Invitrogen,Carlsbad,CA,USA)法提取玉米花药总RNA。利用NanoDrop One检测RNA浓度,用Agilent 2100检测28S/18S以及RIN值,同时用1%琼脂糖凝胶电泳检测所提取RNA的质量及完整性。
1.3 转录组建库及测序
利用Oligo(dT)磁珠富集每个玉米花药样品的mRNA(Illumina,San Diego,CA,USA),加入片段化缓冲液将mRNA随机打断成短片段。将打断后的mRNA反转录成第一链cDNA,随后合成第二链。利用QiaQuick PCR提取试剂盒纯化后进行末端修复并在3'末端加上碱基A,并连接测序接头。筛选大小在300—500 bp的片段进行PCR扩增。文库经Agilent 2100 Bioanalyzer和ABI StepOnePlus Real-Time PCR System质检合格后用Illumina HiSeq 2500进行双末端测序(北京贝瑞和康生物科技有限公司)。
1.4 序列分析
测序获得的原始数据首先过滤掉低质量(Q<30)、接头污染以及位置碱基N含量过高(>5%)的reads,得到clean reads用于后续分析。利用HiSat2软件[29]将过滤后的数据比对到玉米参考基因组B73第四版上(B73 AGPv4,http://ensembl.gramene.org/Zea_mays/ Info/Index),用stringtie软件[30]进行转录本拼接并进行基因表达量估算。基因表达值导入R软件包DESeq2中进行差异基因分析[29]。padj小于0.05及表达差异大于2的基因认为是差异表达的基因(DEG)。根据NCBI的玉米基因注释数据提取差异表达基因的功能信息,利用内部perl脚本从maizeGDB网站上获取差异基因的GO注释。从KAAS网站获取差异表达基因的KEGG途径,利用R软件的clusterProfiler包进行差异基因的GO富集及代谢途径的富集,并对线粒体相关途径进行了分析[32]。
1.5 差异表达基因的半定量验证
分别以87-1“三系”材料的减数分裂前期Ⅰ、四分体期和单核期的花药总RNA为模板,反转录合成cDNA第一链。利用TB GreenTMPremix Ex TaqTMⅡ(TaKaRa)荧光定量试剂盒在CFX96实时定量PCR仪上对转录组数据差异表达的基因、、和进行定量分析。使用Primer3 plus在线设计所有qRT-PCR反应特异引物(表1)。cDNA反转录和qRT-PCR反应体系及反应程序按照PrimeScriptTMRT reagent Kit with gDNA Eraser(TaKaRa)和TB GreenTM Premix Ex TaqTMⅡ(TaKaRa)试剂盒里的说明书的操作进行。每个试验均设3次生物学重复和3次技术重复,利用(2-ΔΔCt)法[33]分析基因表达水平。

表1 差异基因表达分析所用引物
1.6 ATP酶活性测定
根据北京索莱宝科技有限公司(Solarbio)钙镁离子ATP酶检测试剂盒说明书测定C型胞质雄性不育三系减数分裂前期花药的ATP酶活性。称取新鲜样品约0.1 g,加入1 mL试剂一进行冰浴匀浆,9 200 r/min 4℃离心10 min,取上清置于冰上待测。按照手册设置ATP酶酶促反应,水浴10 min,取上清液100 μL,分别加入定磷反应的对照管和测定管,加入定磷剂1 000 μL,混匀,40℃水浴10 min,冷却至室温后利用分光光度计于660 nm处比色。ATP酶活力(U·g-1)=7.5×(A测定管-A对照管)÷(A标准管-A空白管)÷W(样本鲜重,g),计算钙镁离子ATP酶活性。每个材料测定3次。
2 结果
2.1 转录组测序与比对分析
为减少分期播种的环境影响及不同个体间的差异,同一时期至少混合3个不同播期材料及3个单株上的样品用于RNA提取及后续测序分析。所测数据经过严格的质控后,产生5.6亿对双末端序列,共计156.59 Gb的序列数据(表2)。除样品CrT23和CRT23外,Q30比例都超过了90%。PCA分析显示三系前期各有一个样品与相同时期的样品差异较大,在后续比对分析中被剔除;其余不同样品间差异明显,不同样品的生物学重复可聚类在一起。Hisat2比对到参考基因组的序列经过stringtie组装,共得到53 035个Unigene,其中20 017(37.74%)个Unigene为B73基因组中未注释的新基因。
2.2 差异表达基因分析
将差异基因定义为FDR(false discovery rate)<0.05且表达差异倍数在2倍以上的基因(图1)。通过比较87-1三系不同时期及相同时期不同系间的表达谱,获得在三系不同时期或同一材料不同时期之间的显著性差异基因(表3)。87-1不育系在减数分裂中期Ⅰ与前期Ⅰ相比共有121个差异表达基因,其中48个基因个上调,73个基因下调;在四分体时期有329个基因相比中期Ⅰ表现为下调,而与前期Ⅰ相比,差异基因之间表现出显著的变化。87-1保持系在不同发育时期之间差异表达基因的变化趋势与不育系相似,但上调表达的基因数量更多。87-1恢复系在不同发育时期中差异基因数目的变化趋势与保持系和不育系明显不同,中期Ⅰ和前期Ⅰ的差异基因数量与四分体和中期Ⅰ间的差异数量相比变化不大。保持系与不育系的四分体时期出现大量差异表达基因(723上调/409下调),而恢复系与不育系相比在相同时期也有较多差异表达的基因。在87-1三系材料的整个发育时期中有135个基因均存在差异表达。
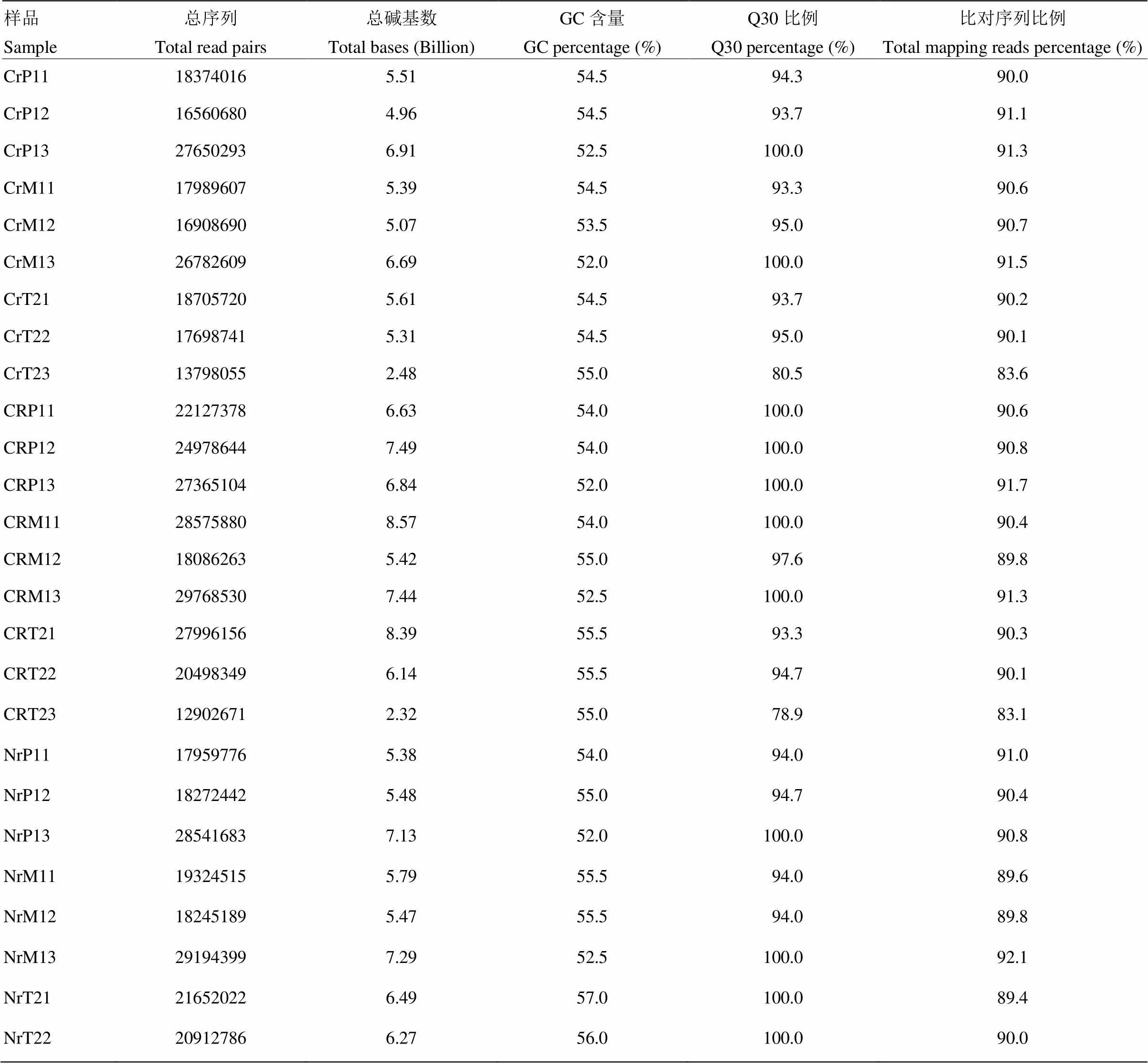
表2 测序数据统计分析
CR:恢复系;Cr:不育系;Nr:保持系;M1:中期Ⅰ;P1:前期Ⅰ;T2:四分体时期(末期Ⅱ);下同。样品名最后一位数字:重复
CR: the restorer lines; Cr: the sterile lines; Nr: the maintainer lines; M1: Metaphase Ⅰ; P1: Prophase Ⅰ; T2: Telophase Ⅱ (tetrad); The same as below. the end digit in sample name: replications
2.3 差异表达基因GO功能分析
将时间轴差异分析结果及两两对比的结果取并集进行分析,共得到5 676个显著的差异表达基因。通过GO分析,有4 273个基因获得4 946个GO注释条目,其中具有3个及以上Unigene的GO条目的基因有2 128个。差异表达基因富集在生物学过程中的有4 231个,细胞组分有4 082个,分子功能有4 085个。富集最多的前81个GO分类如图2所示,细胞组分中富集基因最多的部位是膜的组分(integral component of membrane),其次是核内(nucleus)和质膜上(plasma membrane)的基因,而线粒体中也富集到较多的差异基因;分子功能中富集较多的3个亚类分别是ATP结合、DNA结合及锌离子结合。与转录有关的基因在生物学过程中富集最多,其次还有响应刺激有关的基因以及氧化还原反应有关的基因。时间轴分析的差异基因多富集在与磷酸酶活性相关的GO条目中。87-1恢复系与不育系在减数分裂中期Ⅰ差异表达的基因多富集在花粉发育、配子体发育等生物学过程中。
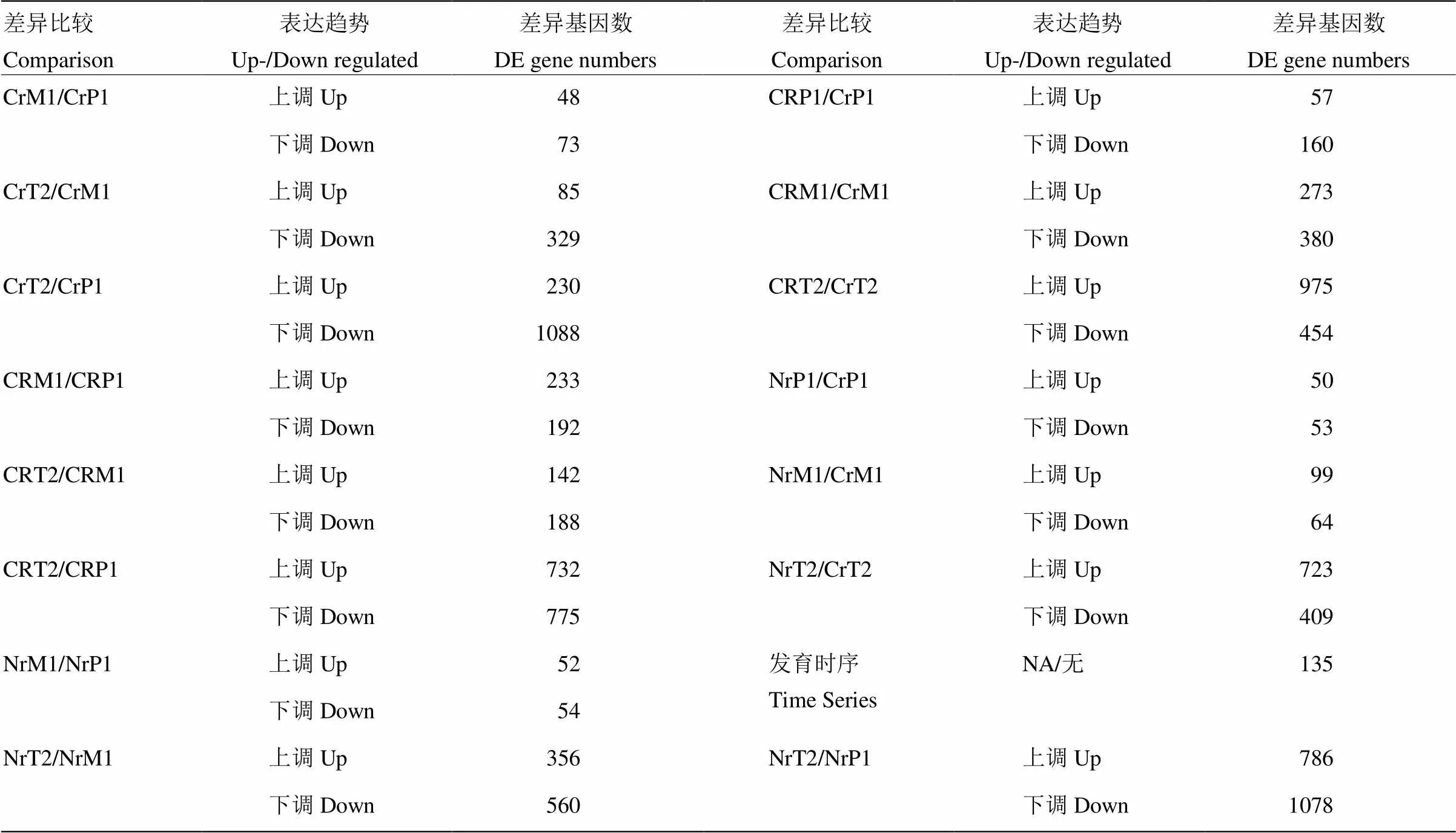
表3 差异基因统计分析
FDR < 0.05 and |log2FC| >= 1
2.4 差异表达基因的KEGG通路分析
通过对87-1三系不同时期两两对比及时间轴差异基因分别进行KEGG分析,共获得37个不同的代谢通路(表4)。在两两对比分析中出现最多次数的途径是角质、软木脂和蜡质合成途径,共出现12次;其次是辅酶Q及其他萜-醌合成、脂肪酸降解和苯丙烷合成途径。在所有途径中,富集基因最多的是碳代谢途径,其次是糖酵解途径和DNA复制途径。
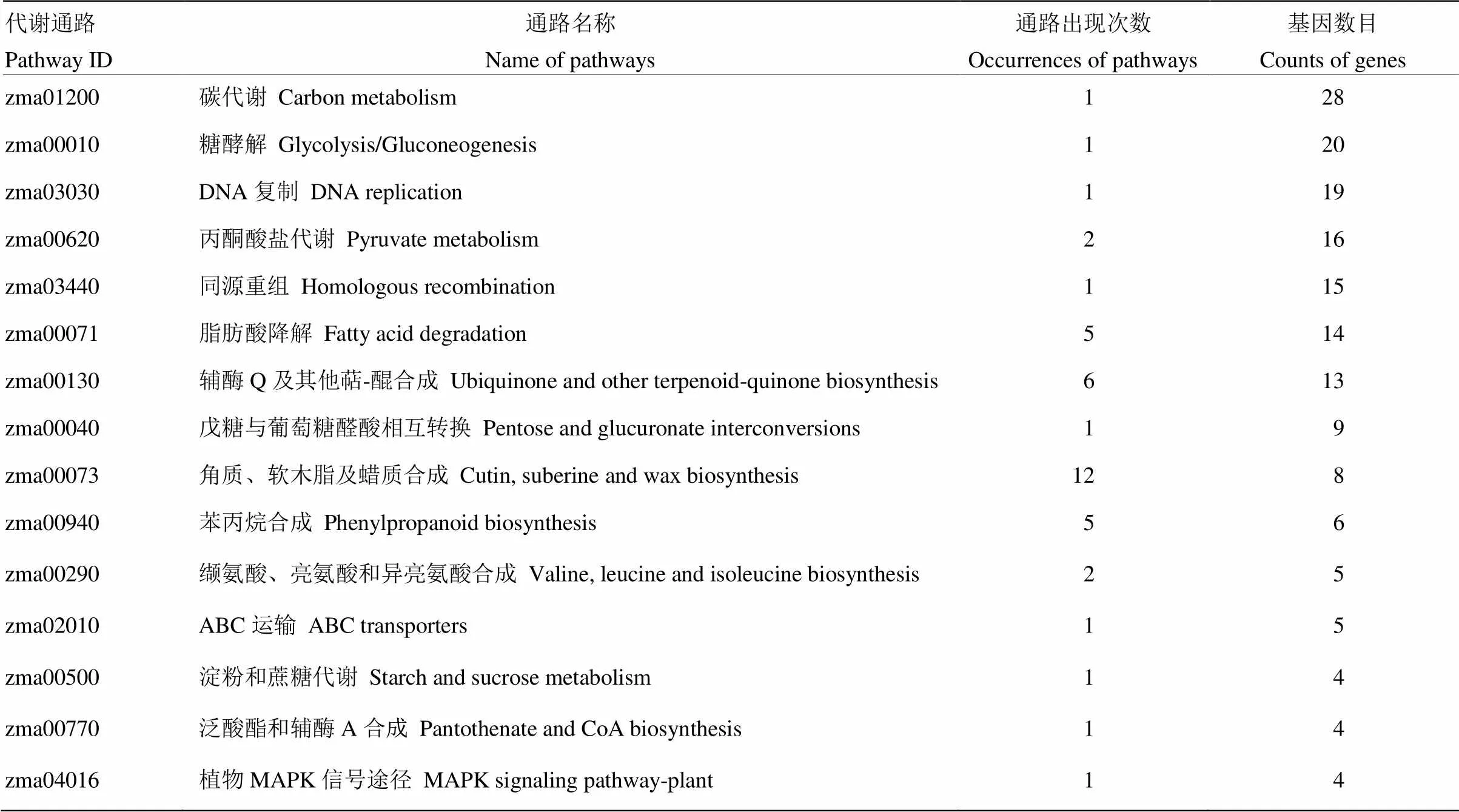
表4 差异表达基因数量最多的15个代谢通路
2.5 线粒体氧化磷酸化途径相关基因的表达分析
线粒体是细胞的能量工厂,而细胞质雄性不育多与线粒体相关,尤其与线粒体内氧化磷酸化途径相关[18,34-35]。玉米中共有187个基因与氧化磷酸化途径有关(KEGG途径zma00190)。将该基因集与差异表达基因取交集,共有13个基因在三系不同发育时期中表达呈显著差异(表5),其中基因和在87-1保持系花药不同发育时期持续上调表达,而在87-1不育系中表达则不存在差异;而在87-1恢复系的花药不同发育时期的表达量与保持系相似(图3-A和图3-B)。87-1保持系与不育系的花药从减数分裂前期到四分体时期的差异表达相比,有4个基因(、、和)显著上调表达;在87-1恢复系中3个基因(、和)的表达趋势与保持系相同,同时有另外3个基因(、和)的表达水平显著提高(表5,图3-C)。值得注意的是,基因在87-1三系的花药发育时期的表达波动较大(图3-D)。上述这些差异表达基因主要富集在氧化磷酸化途径的复合体Ⅱ、复合体Ⅳ和复合体Ⅴ中(图4)。
为验证转录组中差异基因的表达结果,挑选了氧化磷酸化途径中的4个差异表达基因(ComplexⅡ)、(ComplexⅣ)、(ComplexⅤ,F-type ATPase beta)和(ComplexⅤ,inorganic diphosphatase),以为内参基因,通过qRT-PCR技术分析它们在玉米C型胞质不育花药不同发育阶段的表达变化(图5)。结果表明,随着花药的发育表达量逐渐下降,在减数分裂中期Ⅰ时恢复系的表达量约为不育系的2倍;在恢复系中表达趋势与相同,在不育系及保持系中都是先下降再上升,且不育系单核期比中期Ⅰ上升了1倍多;恢复系和保持系中,和从中期Ⅰ到末期Ⅱ的表达趋势相反,从末期Ⅱ到单核期表达趋势相同;在三系材料末期Ⅱ中迅速上升,不育系上升了7倍多,但其表达量仍低于保持系和恢复系。qTR-PCR结果与转录组分析结果基本一致。

A:Zm00001d033552;B:Zm00001d043834;C:Zm00001d009222;D:Zm00001d037576
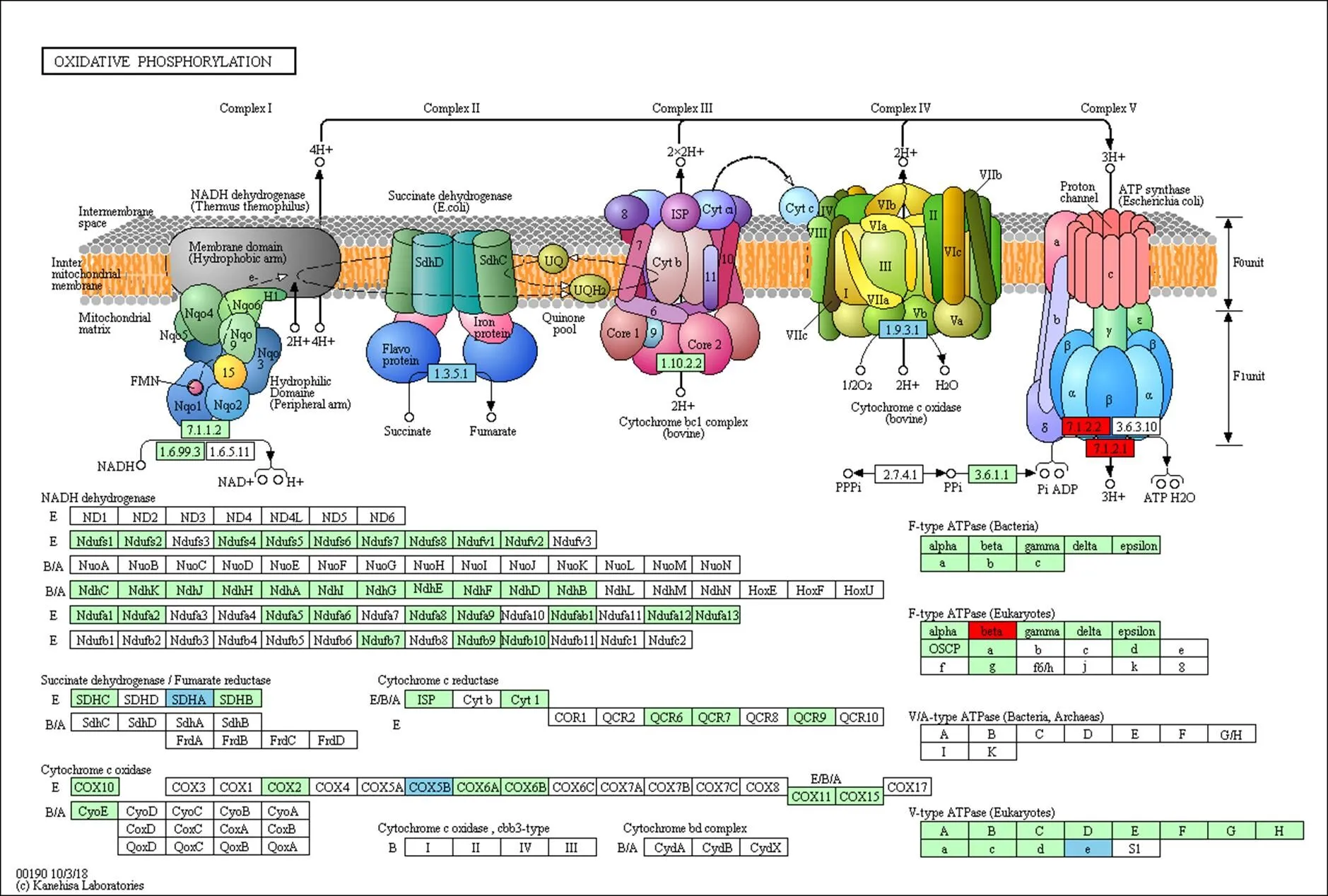
绿底框:物种特异的基因;红底框:上调基因;天蓝底框:下调基因
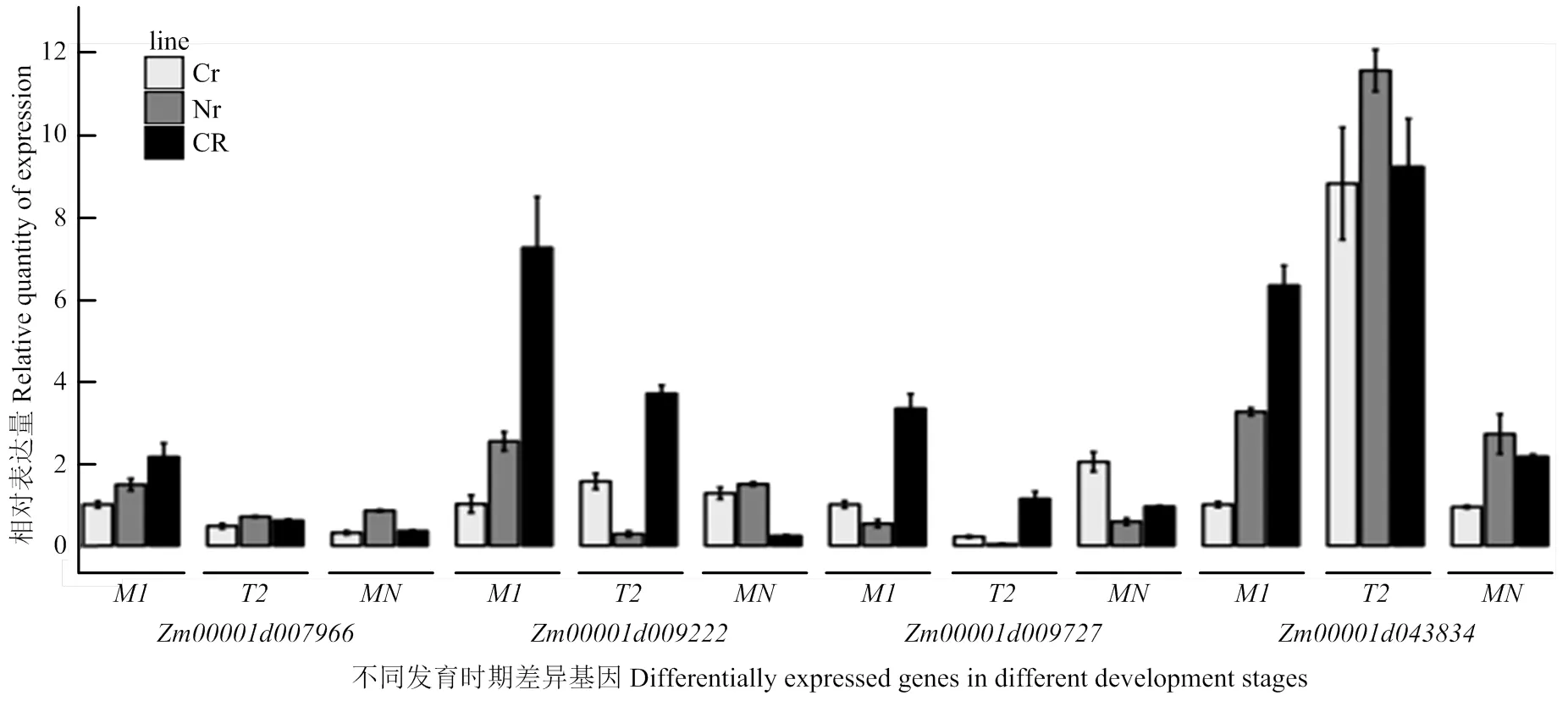
CR:恢复系;Cr:不育系;Nr:保持系,M1:中期1;T2:末期2(tetrad);MN:单核期

表5 线粒体氧化磷酸化途径相关基因的表达差异
2.6 细胞色素P450基因的表达分析
细胞色素P450通过调节许多重要的细胞过程而影响植物的生长与发育,而一些P450基因参与合成植物激素如生长素、赤霉素、细胞分裂素等在植物开花成熟过程中发挥着重要的作用[36-37]。转录组分析不同时期保持系与不育系、恢复系与不育系的比较中发现12个细胞色素P450家族基因的表达存在显著差异(表6)。在减数分裂前期,保持系中与比不育系的表达量要高得多(log2值分别为19.96和17.59),表明不育系中这两个基因几乎不表达或表达量极低;在恢复系中,的表达量恢复到保持系的水平。及至减数分裂中期Ⅰ,这两个基因在不同材料的比较中没有差异,同时保持系与不育系间未出现新的差异表达细胞色素P450家族基因;而在恢复系中新增3个上调及3个下调的细胞色素P450基因。在减数分裂末期2时,前期1的2个差异基因再次出现差异,且表达趋势相同;而在不育系中下调的细胞色素基因在恢复系中都得到了恢复。
2.7 ATP酶活性变化
氧化磷酸化途径是线粒体内重要的代谢途径。玉米C型胞质不育线粒体基因组内发生重复或嵌合的基因(、和)均与该途径相关,其中()位于复合体Ⅳ中,和()位于复合体Ⅴ中。重复的基因打破原有基因间的剂量比例[38-40],而嵌合基因又干扰了正常atp6的生成,并进一步干扰了复合体Ⅴ的功能行使[18],最终引起线粒体ATP酶的活性变化。对“三系”材料减数分裂期间花药ATP酶活性测定结果显示,不育系的ATP酶活性相较于保持系明显低了1个酶活单位(图6),而在恢复系中,ATP酶的活性不但得到恢复,并且比保持系提高了一个酶活单位。
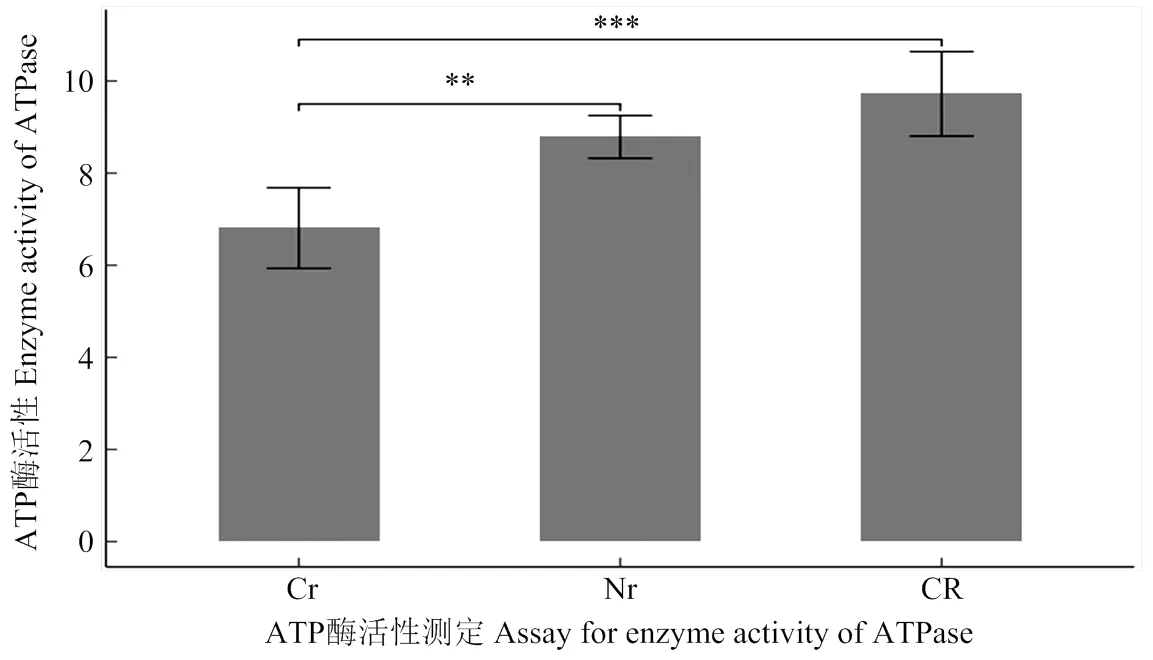
CR:恢复系;Cr:不育系;Nr:保持系;**表示0.01水平差异显著;***表示0.001水平差异显著
3 讨论
3.1 玉米C型胞质雄性不育系败育时期
Lee等[23]通过电镜观察发现玉米C型胞质雄性不育系的花药在四分体时期出现差异,主要为绒毡层细胞出现大量小液泡(类型Ⅰ)或高度液泡化(类型Ⅱ),细胞器如线粒体等正常。进一步观察发现,不育系绒毡层细胞在二分体时期未观察到U小体(Ubisch body),四分体时除出现液泡外,胞质的电子密度亦有降低,小孢子初期开始败育[24]。本研究中发现87-1保持系与不育系在前期仅有103个差异表达基因;而87-1保持系与不育系相比前期Ⅰ至中期Ⅰ的差异表达基因也相对较少,而同一时期,保持系与不育系小孢子发育也没有明显区别,说明这些差异基因可能与后期的小孢子败育无直接关系。87-1不育系的花药从中期Ⅰ到四分体,以及不同材料的花药四分体之间则存在大量的差异表达基因,而前人利用电镜观察到不育系败育的表型相吻合[23-24],因此可以推测引起玉米C胞质雄性不育小孢子败育的关键时期应在减数分裂中期Ⅰ到减数分裂后期Ⅱ之间,而这一阶段正是玉米花药绒毡层细胞二核化的高峰期[41]。Li等[27]曾经对玉米C型胞质雄性不育系及保持系的花粉母细胞及单核期进行了转录组测序,虽然鉴定到一些差异表达的基因,但是这些时期由于不是引起小孢子败育的关键时期,鉴定到的差异表达基因可能与小孢子的后期败育有关。为了筛选出与玉米C型胞质雄性不育小孢子败育与恢复的关键基因,本研究选择了减数分裂前期、减数分裂中期Ⅰ及四分体3个花药发育的关键时期进行了转录组分析[23-24],并从差异不同时期差异基因的变化趋势中推断出基因表达改变的主要时期,为后续表达分析、分子鉴定的取样提供了依据。
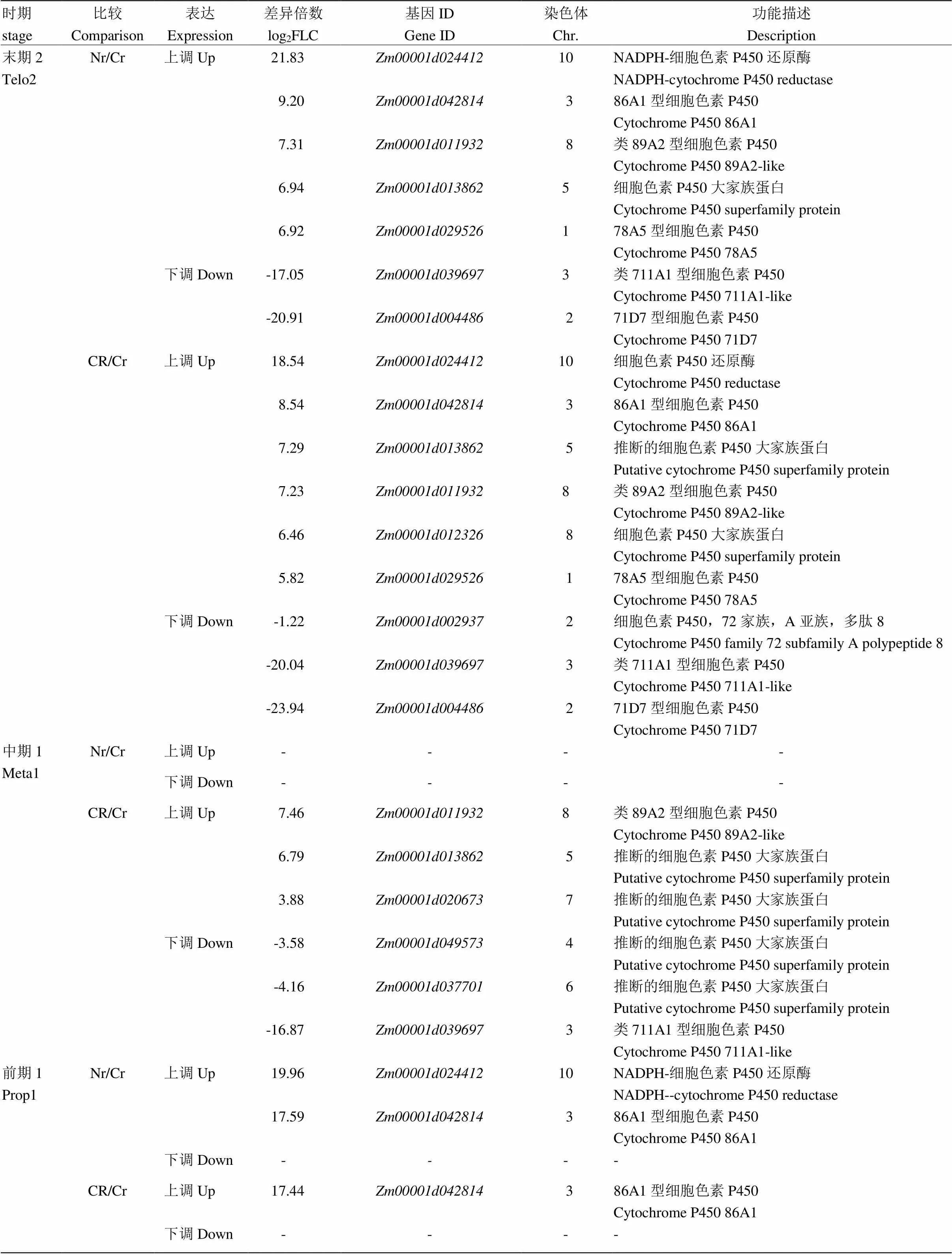
表6 细胞色素P450基因的表达
3.2 玉米C型胞质不育恢复系通过能量补偿恢复育性
前人通过RNA杂交与C型胞质不育系和保持系的线粒体测序分析,发现玉米N胞质与C胞质的线粒体基因组在atp6、atp9和coxⅡ等基因序列间存在差异,其中C胞质线粒体中为嵌合体基因,和具有2个拷贝[21]。线粒体全基因组分析中也发现有其它的嵌合体基因,但与已知的线粒体基因均不同源[22]。尽管这3个基因可能与C胞质不育相关,迄今为止,尚没有分子证据能证明其中的基因为不育基因。Chen等[18]总结了引起细胞质雄性不育的4个模型,分别是毒性假说、能量亏损假说、异常PCD假说及反向调节假说。本研究通过对线粒体基因进行分析,发现上述3个基因组间存在差异的基因在不育系与保持系及恢复系之间的差异表达无显著差异(图7)。由于及是氧化磷酸化途径中复合体Ⅴ的组成部分,为复合体Ⅳ的成分,这些基因微小的表达差异也可引起能量供应的波动,进而造成花粉不育。转录组数据分析结果表明,氧化磷酸化相关基因在87-1不育系中表达较低,而在87-1保持系与恢复系中上调表达(图4,红框基因);而在87-1恢复系中同时还抑制了该途径中另外几个基因的表达(图4,蓝框基因),从而恢复线粒体的能量供求;ATP酶活性测定结果显示ATP酶活性波动趋势(图6)与由转录组数据中相应基因的表达趋势推导的结论一致。氧化磷酸化途径复合体Ⅴ与ATP的合成直接有关,qRT-PCR分析显示,复合体Ⅴ亚基基因在四分体时期达到高峰,且中期Ⅰ中恢复系和保持系中的表达量比不育系高出数倍(图5)。据此推断,玉米C型胞质雄性不育可能是由线粒体能量不足引起的,而恢复基因则可以对不育基因引起的能量亏损进行补偿;同时恢复基因可能通过协调氧化磷酸化途径中不同的复合体亚基,促进整个途径中的反应有序运行,弥补不育基因的有害作用,进而恢复育性。
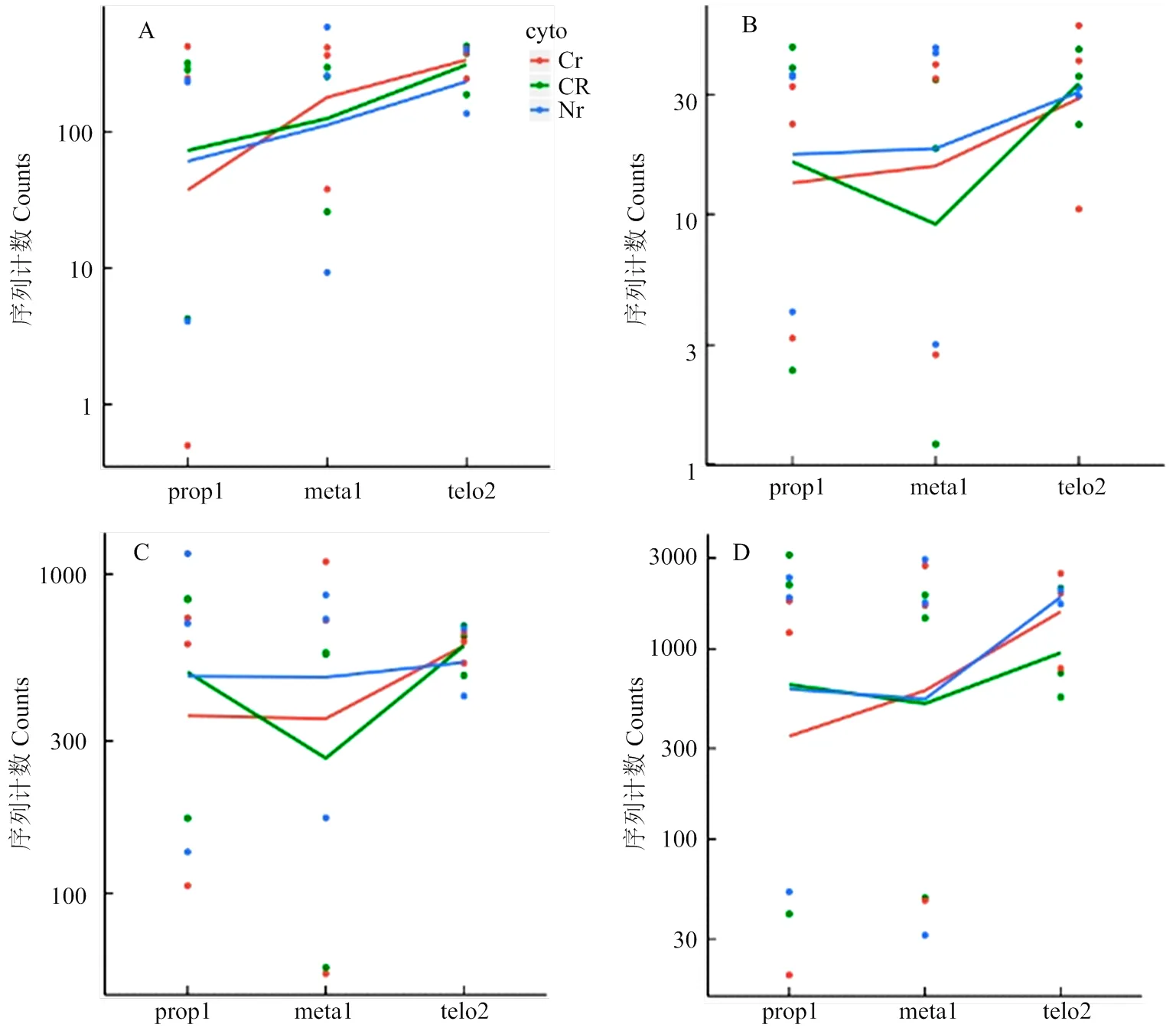
A:atp6c;B:atp9-1;C:cox2-1;D:nad4
3.3 细胞色素P450基因的调控与玉米C型胞质雄性不育相关
植物细胞色素P450基因家族参与多个代谢途径,影响植物的生长发育及开花[37]。过表达的拟南芥植株与野生型相比具有更大的荚果、较短的雄蕊以及较低的育性等[42]。敲除的拟南芥具有更多的叶片[43]。而则参与拟南芥生殖发育[44]。水稻中与花药角质的合成及花粉外壁的形成相关[45]。玉米核不育基因为细胞色素P450类基因[46]。前人研究表明细胞色素P450家族的基因与雄性器官的发育和育性有关。本研究发现减数分裂前期Ⅰ时不育系相比保持系,细胞色素P450基因与表达量极低,恢复系通过恢复其中一个细胞色素P450基因使得减数分裂正常向后进行。随后恢复系中通过恢复更多的细胞色素P450基因并抑制该家族中不需要的基因保证绒毡层细胞的正常功能最终恢复花粉的育性。由此推测,不育基因可能影响到细胞色素P450基因的表达,而恢复基因通过早期恢复相应细胞色素P450基因的表达,进而级联恢复后续发育所需的该家族基因,保证了细胞功能的正常行使,从而使育性得以恢复。
4 结论
玉米C型胞质不育形成的关键时期可能为减数分裂中期Ⅰ与减数分裂后期Ⅱ之间;玉米C型胞质不育机理可能是能量亏损假说,恢复基因通过直接或间接作用进行能量补偿而恢复育性。
[1] Dewey R, Levings C S, Timothy D H. Novel recombinations in the maize mitochondrial genome produce a unique transcriptional unit in the texas male-sterile cytoplasm., 1986, 44(3): 439-449.
[2] Peng X, Wang K, Hu C, Zhu Y, Wang T, Yang J, Tong J, Li S, Zhu Y. The mitochondrial gene orfH79 plays a critical role in impairing both male gametophyte development and root growth in CMS-Honglian rice., 2010, 10(1): 125.
[3] Wang Z, Zou Y, Li X, Zhang Q, Chen L, Wu H, Su D, Chen Y, Guo J, Luo D, Long Y, Zhong Y, Liu Y G. Cytoplasmic male sterility of rice with Boro II cytoplasm is caused by a cytotoxic peptide and is restored by two related PPR motif genes via distinct modes of mRNA silencing., 2006, 18(3): 676-687.
[4] Luo D, Xu H, Liu Z, Guo J, Li H, Chen L, Fang C, Zhang Q, Bai M, Yao N, Wu H, Wu H, Ji C, Zheng H, Chen Y, Ye S, Li X, Zhao X, Li R, Liu Y G. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice., 2013, 45(5): 573-577.
[5] Singh M. Suppression of cytoplasmic male sterility by nuclear genes alters expression of a novel mitochondrial gene region., 1991, 3(12): 1349-1362.
[6] Brown G G. Unique aspects of cytoplasmic male sterility and fertility restoration in., 1999, 90(3): 351-356.
[7] Uyttewaal M, Arnal N, Quadrado M, Martin-Canadell A, Vrielynck N, Hiard S, Gherbi H, Bendahmane A, Budar F, Mireau H. Characterization ofpentatricopeptide repeat proteins encoded by the fertility restorer locus for Ogura cytoplasmic male sterility., 2008, 20(12): 3331-3345.
[8] Cui X, Wise R P, Schnable P S. Thenuclear restorer gene of male-sterile T-cytoplasm maize., 1996, 272(5266): 1334-1336.
[9] Fujii S, Toriyama K. Suppressed expression of retrograde- regulated male sterility restores pollen fertility in cytoplasmic male sterile rice plants., 2009, 106(23): 9513-9518.
[10] Itabashi E, Iwata N, Fujii S, Kazama T, Toriyama K. The fertility restorer gene,, for lead rice-type cytoplasmic male sterility of rice encodes a mitochondrial glycine-rich protein., 2011, 65(3): 359-367.
[11] Kitazaki K, Arakawa T, Matsunaga M, Yui-Kurino R, Matsuhira H, Mikami T, Kubo T. Post-translational mechanisms are associated with fertility restoration of cytoplasmic male sterility in sugar beet ()., 2015, 83(2): 290-299.
[12] Bentolila S, Alfonso A A, Hanson M R. A pentatricopeptide repeat-containing gene restores fertility to cytoplasmic male-sterile plants., 2002, 99(16): 10887-10892.
[13] Brown G G, Formanová N, Jin H, Wargachuk R, Dendy C, Patil P, Laforest M, Zhang J, Cheung W Y, Landry B S. The radishrestorer gene of Ogura cytoplasmic male sterility encodes a protein with multiple pentatricopeptide repeats., 2003, 35(2): 262-272.
[14] Koizuka N, Imai R, Fujimoto H, Hayakawa T, Kimura Y, Kohno-Murase J, Sakai T, Kawasaki S, Imamura J. Genetic characterization of a pentatricopeptide repeat protein gene,, that restores fertility in the cytoplasmic male-sterile Kosena radish., 2003, 34(4): 407-415.
[15] Hu J, Wang K, Huang W, Liu G, Gao Y, Wang J, Huang Q, Ji Y, Qin X, Wan L, Zhu R, Li S, Yang D, Zhu Y. The rice pentatricopeptide repeat proteinrestores fertility in Hong-Lian cytoplasmic male-sterile lines via a complex with the glycine-rich protein GRP162., 2012, 24(1): 109-122.
[16] Tang H, Luo D, Zhou D, Zhang Q, Tian D, Zheng X, Chen L, Liu Y G. The rice restorerfor wild-abortive cytoplasmic male sterility encodes a mitochondrial-localized PPR protein that functions in reduction of WA352 transcripts., 2014, 7(9): 1497-1500.
[17] Huang W, Yu C, Hu J, Wang L, Dan Z, Zhou W, He C, Zeng Y, Yao G, Qi J, Zhang Z, Zhu R, Chen X, Zhu Y. Pentatricopeptide-repeat family protein RF6 functions with hexokinase 6 to rescue rice cytoplasmic male sterility., 2015, 112(48): 14984-14989.
[18] Chen L, Liu Y G. Male sterility and fertility restoration in crops., 2014, 65(1): 579-606.
[19] 陈伟程, 罗福和, 季良越. 玉米C型胞质雄花不育的遗传及其在生产上的应用. 作物学报, 1979, 5(4): 21-28.
Chen W C, Luo F H, Ji L Y. Some genetic aspects of the C-type cytoplasmic male-sterility in maize and its use in breeding., 1979, 5(4): 21-28. (in Chinese)
[20] 汤继华, 刘宗华, 陈伟程, 胡彦民, 季洪强, 季良越. 玉米C型胞质不育恢复主基因SSR标记. 中国农业科学, 2001, 34(6): 592-596.
Tang J H, Liu Z H, Chen W C, Hu Y M, Ji H Q, Ji L Y. The SSR markers of the main restorer genes for CMS-C cytoplasmic male sterility in maize., 2001, 34(6): 592-596. (in Chinese)
[21] Dewey R E, Timothy D H, Levings C S. Chimeric mitochondrial genes expressed in the C male-sterile cytoplasm of maize., 1991, 20(6): 475-482.
[22] Allen J O, Fauron C M, Minx P, Roark L, Oddiraju S, Lin G N, Meyer L, Sun H, Kim K, Wang C, Du F, Xu D, Gibson M, Cifrese J, Clifton S W, Newton K J. Comparisons among two fertile and three male-sterile mitochondrial genomes of maize., 2007, 177(2): 1173-1192.
[23] Lee S L, Gracen V E, Earle E D. The cytology of pollen abortion in C-cytoplasmic male-sterile corn anthers., 1979, 66(6): 12.
[24] 陈伟程, 李桂珍. 玉米C型胞质雄性不育系花粉败育的细胞学研究. 华北农学报, 1987, 2(1): 1-6.
Chen W C, Li G Z. A cytological study in pollen abortion in C-cytoplasmic male-sterile corn (, L.)., 1987, 2(1): 1-6. (in Chinese)
[25] Liu Q, Lan Y, Wen C, Zhao H, Wang J, Wang Y. Transcriptome sequencing analyses between the cytoplasmic male sterile line and its maintainer line in Welsh onion (L.)., 2016, 17(7): 1058.
[26] Liu C, Ma N, Wang P Y, Fu N, Shen H L. Transcriptome sequencing and De Novo analysis of a cytoplasmic male sterile line and its near-isogenic restorer line in Chili pepper (L.)., 2013, 8(6): e65209.
[27] Li C, Zhao Z, Liu Y, Liang B, Guan S, Lan H, Wang J, Lu Y, Cao M. Comparative transcriptome analysis of isonuclear-alloplasmic lines unmask key transcription factor genes and metabolic pathways involved in sterility of maize CMS-C., 2017, 5: e3408.
[28] Ma J, Skibbe D S, Fernandes J, Walbot V. Male reproductive development: gene expression profiling of maize anther and pollen ontogeny., 2008, 9(12): R181.
[29] Kim D, Langmead B, Salzberg S L. HISAT: a fast spliced aligner with low memory requirements., 2015, 12(4): 357-360.
[30] Pertea M, Pertea G M, Antonescu C M, Chang T C, Mendell J T, Salzberg S L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads., 2015, 33(3): 290-295.
[31] Love M I, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2., 2014, 15(12): 550.
[32] Yu G, Wang L G, Han Y, He Q Y. ClusterProfiler: an R package for comparing biological themes among gene clusters., 2012, 16(5): 284-287.
[33] Livak K J, Schmittgen T D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCTmethod., 2001, 25(4): 402-408.
[34] Hu J, Huang W C, Huang Q, Qin X J, Yu C C, Wang L L, Li S Q, Zhu R S, Zhu Y G. Mitochondria and cytoplasmic male sterility in plants., 2014, 19(B): 282-288.
[35] Touzet P, Meyer E H. Cytoplasmic male sterility and mitochondrial metablolism in plants., 2014, 19(B): 166-171.
[36] Werck-Reichhart D, Feyereisen R. Cytochromes P450: a success story., 2000, 1(6): reviews3003.1-3003.9.
[37] Xu J, Wang X Y, Guo W Z. The cytochrome P450 superfmily: Key players in plant development and defense., 2015, 14(9): 1673-1686.
[38] Qian W F, Zhang J Z. Gene dosage and gene duplicability., 2008, 179(4): 2319-2324.
[39] Spadafora N, Perrotta L, Nieuwland J, Albani D, Bitonti B M, Herbert R J, Doonan J H, Marchbank A M, Siciliano I, Grønlund A L, Francis D, Rogers H J. Gene dosage effect of WEE1 on growth and morphogenesis fromexplants., 2012, 110(8): 1631-1639.
[40] Chang N, Sun Q Q, Li Y Q, Mu Y J, Hu J L, Feng Y, Liu X M, Gao H B. PDV2 has a dosage effect on chloroplast division in., 2017, 36(3): 471-480.
[41] Kelliher T, Walbot V. Emergence and patterning of the five cell types of theanther locule., 2011, 350(1): 32-49.
[42] Fang W, Wang Z, Cui R, Li J, Li Y. Maternal control of seed size by EOD3/CYP78A6 in., 2012, 70: 929-939.
[43] Wang J W, Schwab R, Czech B, Mica E, Weigel D. Dual effects of miR156-targeted SPL genes and CYP78A5/KLUH on plastochron length and organ size in., 2008, 20: 1231-1243.
[44] Sotelo-Silveira M, Cucinotta M, Chauvin A L, Chavez Montes R A, Colombo L, Marsch-Martinez N, de Folter S. Cytochrome P450 CYP78A9 is involved inreproductive development., 2013, 162: 779-799.
[45] Li H, Pinot F, Sauveplane V, Werck-Reichhart D, Diehl P, Schreiber L, Franke R, Zhang P, Chen L, Gao Y W, Liang W Q, Zhang D B. Cytochrome P450 family member CYP704B2 catalyzes the omega-hydroxylation of fatty acids and is required for anther cutin biosynthesis and pollen exine formation in rice., 2010, 22: 173-190.
[46] Djukanovic V, Smith J, Lowe K, Yang M Z, Gao H R, Jones S, Micholson M G, West A, Lape J, Bidney D, Falco S C, Jantz D, Lyznik L A. Male-sterile maize plants produced by targeted mutagenesis of the cytochrome P450-like gene (MS26) using a re-designed I-CreI homeing endonuclease., 2013, 76: 888-899.
Comparative transcriptome analysis among the three line of cytoplasmic male sterility in maize
XUE Yadong1, YANG Lu1, YANG Huili1, LI Bing1, LIN Yanan1, ZHANG Huaisheng1, GUO Zhanyong1, TANG Jihua1,2
(1College of Agronomy, Henan Agricultural University/Key Laboratory of Wheat and Maize Crops Science, Zhengzhou 450002;2Hubei Collaborative Innovation Center for Grain Industry, Yangtze University, Jingzhou 434025, Hubei)
【Objective】It is one of the most efficient ways to utilize cytoplasmic male sterile (CMS) lines in hybrid seed production, which could improve the purity of seeds, reduce the cost in creating hybrid seeds and enhance the competitiveness of Chinese seed companies. The comparative transcriptome analysis of the anthers at different development stages from the CMS line, the maintainer line and the restorer line (the three lines) were performed in order to understand the mechanism of sterility and restoration of CMS-C in maize, and also to elucidate the regulation network between the restorer gene and the sterile gene, which will provide the fundamental basis for the employment of maize CMS in hybrid seed production.【Method】The transcriptome sequencing was carried out on the anthers at the prophaseⅠ, the metaphaseⅠand the tetrad stage from the three lines based on the elite inbred line Yu87-1. Method of comparative analysis was used to deal with all the transcripts by the tools such as hisat2, ballgown and DESeq2, and to predict genes involved in the regulation network between the sterile gene and the restorer gene, between the different development stages and through the development time series. qRT-PCR was used to verify the differentially expressed genes. The activity of ATPase was quantified with by the spectrophotometric method for the verification of the putative hypothesis.【Result】Transcriptome sequencing totally produced 156.59 Gb sequence data. After mapping and assembling, 53035 Unigenes were obtained. A total of 5676 differentially expressed (DE) genes were identified from the pairwise comparisons (except for comparisons between the restorer lines and the maintainer lines) in the anthers at the different stages from the three lines. Of those, 4705 DE genes between the comparisons of the development stages, 2693 DE genes between the comparisons of the different lines and 135 DE genes related to the time series. The GO molecular functional analysis showed that the genes related to ATP binding, DNA binding and zinc ion binding were highly enriched, in cell component analysis, genes located in integral component of membrane, nucleus and plasma membrane were enriched, and in biological process, genes involved in DNA-templated transcription, regulation of transcription, oxidation-reduction process and primary metabolic process were enriched. KEGG pathway analysis indicated that the oxidative phosphorylation pathways, the carbon metabolism pathways and glycolysis pathways were mostly enriched. Compared to the maintainer lines, several genes involving in the oxidative phosphorylation pathways were significantly down-regulated in the sterile lines, while those down-regulated genes were recovered, besides other genes in the same pathways were also coordinately regulated. The expression trend determined by qRT-PCR on the selected DE genes was in accordance with that in the transcriptome data. The enzyme activity results show that the activity of ATPase in the sterile line was greatly reduced compared to the maintainer line, while in the restorer line, the activities the ATPase were restored due to the existence of the restorer gene.【Conclusion】The onset of the changes in the gene expression caused by the sterile gene in the anthers of CMS-C maize may happen after metaphaseⅠ and before telophase Ⅱ in meiosis before visible phenotype occurred. The energy deficiency model may account for the mechanism of the sterility in maize CMS-C, and the energy requirements were compensated by the restorer gene through direct or indirect manner.
; cytoplasmic male sterility; transcriptome; differentially expressed gene; regulation network
2018-12-10;
2019-02-14
国家自然科学基金(31471504)
薛亚东,E-mail:yadongxue@henau.edu.cn。通信作者汤继华,Tel:0371-56990336;E-mail:tangjihua1@163.com
10.3864/j.issn.0578-1752.2019.08.002
(责任编辑 李莉)
