PI3K/AKT/mTOR信号通路在Burkitt淋巴瘤中的研究进展
2019-05-07孙媛媛张明智
孙媛媛,张明智
0 前言
Burkitt淋巴瘤是一种与Epstein Barr病毒(EBV)、人类免疫缺陷病毒(HIV)的感染有关的高度侵袭性非霍奇金淋巴瘤,是人类生长最快的肿瘤[1]。分为地方型、散发型与免疫缺陷相关型三种类型,在流行病学及miRNA上有各自特点[2]。该病主要发生在儿童及青少年,少数发生于成年人,经短期、强化治疗,如CODOX-M/IVAC方案治疗,疗效显著[3],但感染并发症、中枢受累等因素可能导致患者预后差[4-5]。短期、强效的化疗方案,不良反应强,故对于晚期、耐药、体质较差的患者,寻找精准、高效、低毒的靶向药物显得尤为重要。PI3K/AKT/mTOR信号通路在细胞的生长、分化、代谢、生存以及增殖等方面发挥重要作用[6],其在Burkitt 淋巴瘤中激活,通过对该通路的深入研究,为Burkitt淋巴瘤的靶向治疗提供理论基础。近几年,很多针对该信号通路的新药已进入实验、临床研究,以期为淋巴瘤患者提供更精准的治疗。现就PI3K/AKT/mTOR信号通路为基础的Burkitt淋巴瘤研究进展作一总结。
1 PI3K/AKT/mTOR信号通路的作用机制
磷脂酰肌醇-3激酶(phosphatidylinositol 3-kinase,PI3K)根据结构和功能分为Ⅰ、Ⅱ和Ⅲ型,其中Ⅰ型在肿瘤细胞中广泛突变。Ⅰ型PI3K是由调节亚基P53和催化亚基P110组成的异源二聚体,又分为ⅠA、ⅠB两型,ⅠA型催化亚基由P110α、P110β和P110δ组成,可被细胞表面酪氨酸激酶激活,ⅠB型催化亚基为P110γ,可被G-蛋白偶联受体激活。被激活的PI3K使其底物3,4二磷酸磷脂酰肌醇(PIP2)磷酸化为3,4,5三磷酸磷脂酰肌醇(PIP3),进而使信号通路活化。PI3K的下游效应分子:蛋白激酶B,又名AKT,可通过磷酸化作用,促进细胞生长,抑制细胞凋亡。而AKT可直接活化哺乳动物雷帕霉素靶蛋白(mammal target of rapamycin, mTOR),mTOR包括两种不同的多分子复合物,mTORC1和mTORC2,其在肿瘤细胞中常过度活化。mTORC1被AKT激活后使真核细胞启动因子4E结合蛋白1(4EBP1)和核糖体蛋白p70S6K磷酸化[7]。磷酸化的4EBP1与真核细胞翻译抑制因子(eIF-4E)分离失活,分离的eIF-4E与翻译分子结合,启动蛋白质的翻译;同样,磷酸化的p70S6K也可以促进蛋白质的合成,见图1。PI3K/AKT/mTOR信号通路参与蛋白质的合成,在细胞的生长、分化、代谢、生存以及增殖等方面发挥重要作用[8]。目前,一些关于此通路抑制剂的研究,如AKT抑制剂MK2206的Ⅱ期临床试验,结果显示患者获益:8例中,2例获得完全缓解,6例获得部分缓解,中位缓解时间为5~8月[9]。所以,这就要求我们根据实验、临床研究,探讨PI3K/AKT/mTOR信号通路在Burkitt淋巴瘤中的活化机制,应用抑制剂抑制该通路活化,为治疗复发难治的Burkitt淋巴瘤患者寻找新的方法。
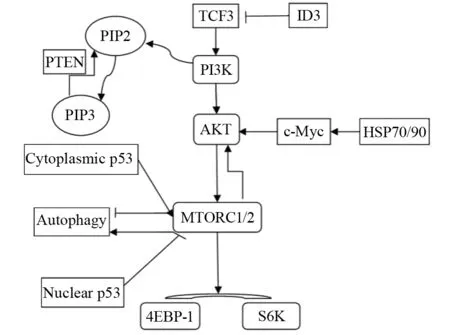
图1 PI3K/AKT/mTOR信号通路与其他信号在Burkitt淋巴瘤相互作用图解Figure1 Schematic of interaction between PI3K/AKT/mTOR signal pathway and other signals in Burkitt lymphoma
2 PI3K/AKT/mTOR信号通路在Burkitt淋巴瘤中的活化
研究发现,PI3K在肿瘤细胞中主要通过扩增及增加复制导致翻译增加、通路活化,而基因突变少见[8]。在Burkitt淋巴瘤中,PI3K/AKT/mTOR信号通路中任何分子异常都可能导致通路的活化。Sander等[10]通过免疫印迹实验(Western blot)发现AKT第437位上丝氨酸和p70S6K第389位上苏氨酸磷酸化,并且应用PI3K抑制剂LY-294002可逆转磷酸化的激酶,结果证实了PI3K通路在Burkitt淋巴瘤中活化。Schmitz等[11]应用高通量的RNA测序和RNA干涉筛选发现在散发型Burkitt淋巴瘤中,70%存在转录因子TCF3或其负调节因子ID3突变,而TCF3的突变可引起PI3K通路的活化。第10号染色体缺失性磷酸酶-张力蛋白同源性基因(phosphatase and tension homology deleted on chromosome ten,PTEN)是一种肿瘤抑制基因,通过对PIP3去磷酸化,对AKT起到负调节作用[8]。PTEN的缺失,可使PI3K/AKT/mTOR信号通路活化,进而导致肿瘤的发生,常常提示疾病预后不良[12]。Pfeifer等[13]的研究中,PTEN的缺失与PI3K/AKT/mTOR和c-Myc信号通路活化有关,用全PI3K抑制剂LY294002处理弥漫大B细胞淋巴瘤患者样本及淋巴瘤细胞系,发现细胞活性降低,样本分析显示55%的生发中心来源的弥漫大B细胞淋巴瘤存在PTEN缺失,这为生发中心来源的Burkitt淋巴瘤的发病机制和治疗方法提供了新的研究方向。
研究已证实PI3K/AKT/mTOR信号通路在Burkitt淋巴瘤中活化,故可通过应用PI3K/AKT/mTOR信号通路抑制剂靶向治疗Burkitt淋巴瘤。PI3K抑制剂主要分三种类型:全Ⅰ型PI3K抑制剂,选择性PI3K亚型抑制剂,全Ⅰ型PI3K和mTOR双元抑制剂[14]。早期的全Ⅰ型PI3K抑制剂,如LY294002 或渥曼青霉素,因脱靶作用等,现临床较少应用。新型全Ⅰ型PI3K抑制剂,例如:BKM120(buparlisib)、SAR245408和BAY 80-6946显示出较少的脱靶作用及较好的耐受性。目前,多种选择性PI3K亚型抑制剂已进入临床研究,例如:p110δ抑制剂idelalisib、PI3Kγδ抑制剂IPI-145和PI3K-αβ抑制剂Rigosertib等,对各种类型淋巴瘤显示出较好的疗效[15]。第一代AKT抑制剂哌立福辛,因临床效果一般,且存在细胞毒性,现已没有针对此药物的临床试验。第二代AKT抑制剂MK-2206,不论是临床前淋巴瘤细胞系研究,还是对患者的临床研究,都有显著效果。mTORC1抑制剂包括依维莫司、替西罗莫司等,然而,mTORC1抑制可通过mTORC2的负反馈作用使PI3K通路活化,进而使AKT磷酸化[8],故单一的mTORC1抑制剂可能因AKT的磷酸化而使患者复发或耐药。李纯团[16]发现,NVPBEZ235可通过同时抑制PI3K、mTOR的活性,引起下游信号分子AKT、PRS6磷酸化水平下降,PI3K/AKT/mTOR信号通路传导减弱,进而起到抗Burkitt淋巴瘤的作用。我们还发现其他药物通过抑制PI3K/AKT/mTOR信号通路,起到治疗Burkitt淋巴瘤、其他类型淋巴瘤及实体瘤的作用,如PI3Kαγδ抑制剂GDC-0332、PI3K-αδ抑制剂Pictilisib[17]、PI3K-α抑制剂BYL719[18]以及PI3K/mTOR抑制剂GDC-0084[19]、Apitolisib[20]等。
细胞中存在多种信号通路,各通路间可通过正负调节相互联系,一条通路被抑制,可能引起另一通路的激活,故单一的PI3K/AKT/mTOR信号通路的靶向治疗易导致患者复发、耐药,这就需要我们寻找针对不同靶点的多重抑制剂。Huang等[21]发现在Burkitt淋巴瘤CA46细胞中,P-AKT高表达,而黄岑苷可以使P-AKT和P-mTOR下调并促进Burkitt淋巴瘤细胞凋亡。Lim等通过体外实验得出,漆黄素通过靶向PI3K和mTOR通路诱导Burkitt淋巴瘤Raji细胞凋亡[22]。目前,应用PI3K/AKT/mTOR信号通路抑制剂对各种类型淋巴瘤进行靶向治疗已进入实验、临床研究,但少有针对Burkitt淋巴瘤的临床试验,期待未来有更多的临床研究投入,为成年、晚期、耐药的Burkitt淋巴瘤患者带来治疗希望。近几年,研究发现PI3K/AKT/mTOR通路受多种因素影响,除了PTEN外,PI3K/AKT/mTOR通路与c-Myc基因、自噬等在Burkitt淋巴瘤中相互作用受到越来越多研究者的重视,通过实验及临床研究,探讨相互关系及作用机制,为患者提供了新的治疗思路。
3 c-Myc表达和PI3K/AKT/mTOR信号通路在Burkitt淋巴瘤中的关系
在所有类型的Burkitt淋巴瘤中,c-Myc通过与免疫球蛋白基因座易位,导致自身过度表达,其表达的蛋白分子是参与细胞分化与凋亡等活动的细胞转录因子。c-Myc一方面通过促进细胞增殖,抑制细胞分化的方式,促进肿瘤发生,但同时也促进细胞凋亡[10]。在Burkitt淋巴瘤细胞中,PI3K/AKT/mTOR信号通路可促进热休克蛋白70和90(HSP70/90)的表达,这些分子伴侣表达上调,参与调节c-Myc的稳定性[23],相应地,c-Myc也可调节mTOR的活性[24],见图1。Park等利用大鼠,将c-Myc插入免疫球蛋白基因促使其过度表达,但未诱导形成Burkitt淋巴瘤模型[25]。Sander等指出单一的c-Myc过度表达并不足以导致肿瘤的发生,并通过MYC过度表达和PI3K通路的激活,在转基因大鼠上诱导出类似人类Burkitt淋巴瘤的生发中心B细胞来源的淋巴瘤,并且在来源于生发中心B细胞肿瘤中发现c-Myc和P110共表达[26]。虽然c-Myc具有促进细胞增殖,抑制细胞分化的作用,但因其也可促进细胞凋亡,所以,c-Myc过度表达联合PI3K/AKT/mTOR信号通路激活才可导致肿瘤的发生。Granato等[24]通过研究槲皮素对Burkitt淋巴瘤的作用,发现可以通过减少c-Myc表达和PI3K/AKT/mTOR通路活化抑制Burkitt淋巴瘤。上述实验证实了c-Myc与PI3K/AKT/mTOR信号通路在Burkitt淋巴瘤的发生上相互作用,促进肿瘤的生成,成为Burkitt淋巴瘤治疗的又一新靶点。
4 自噬和PI3K/AKT/mTOR信号通路在Burkitt淋巴瘤治疗中的作用
与c-Myc作用机制不同,自噬是由自噬相关基因调控的一种细胞死亡过程,与PI3K/AKT/mTOR信号通路、p53(抑癌基因)等有关。p53对细胞自噬具有双重调控作用,一方面,胞质中的p53通过激活mTOR/AMP激酶通路抑制自噬,另一方面,胞核内的p53则通过抑制mTOR和使Beclin与Bcl-2解离,结合Ⅲ型PI3K,促进自噬。当胰岛素或生长因子与其受体结合后,激活mTORC1,促进自噬。PI3K/AKT/mTOR信号通路与细胞自噬过程密切相关,故可通过一些药物作用于该信号通路,促进细胞自噬性死亡,杀死肿瘤细胞,从而治疗Burkitt淋巴瘤。Dong等将替西罗莫司与丙戊酸共同作用于Burkitt淋巴瘤细胞系,发现通过抑制mTOR通路和MYC癌基因蛋白可促进细胞自噬死亡[27]。Granato等[24]发现,槲皮素可通过抑制mTOR通路,引发Burkitt淋巴瘤细胞自噬。而雷帕霉素和地塞米松联合应用,可通过mTOR/P70S6K通路抑制Raji细胞(Burkitt淋巴瘤细胞)的活性[28-29]。有研究报道,肿瘤细胞在缺氧等微环境中可启动保护性自噬,提高生存能力,而放化疗可能导致肿瘤细胞通过增强自噬来减少伤害,与肿瘤的耐药机制有关。Maclean等将自噬抑制剂氯喹作用与Burkitt淋巴瘤小鼠模型,使p53依赖的细胞凋亡[30]。故我们可以通过应用自噬抑制剂,增强肿瘤细胞对放化疗的敏感度,提高治疗效果。
综上可知,自噬对Burkitt淋巴瘤治疗具有双重作用,一方面,通过PI3K/AKT/mTOR信号通路,激活自噬,促进肿瘤细胞的自噬性死亡,另一方面,又能通过自噬抑制剂,抑制自噬,增强肿瘤细胞对放化疗的敏感度。故我们要充分研究不同药物的作用机制,利用PI3K/AKT/mTOR信号通路与自噬的关系,提高成年、晚期、耐药的Burkitt淋巴瘤患者治疗效果,减少不良反应,减轻患者痛苦。
5 展望
研究已证实PI3K/AKT/mTOR信号通路在Burkitt淋巴瘤中呈激活状态,该通路的抑制剂对Burkitt淋巴瘤有抑制作用。但Burkitt淋巴瘤发病率较低,目前针对该疾病的PI3K/AKT/mTOR通路靶向治疗的临床试验较少,期待未来有更多的临床研究投入,同时加强对PTEN、c-Myc、自噬与PI3K/AKT/mTOR信号通路的多重抑制的研究,设计与单克隆抗体或其他通路抑制剂的联合用药。目前正在研究中的其他基因如:ID3、TCF3、CCND3、GNA13和SMARCA4等,在Burkitt淋巴瘤中突变率高,这些突变基因将成为新的治疗靶点[31-33]。此外,我们更需要增加新技术,如二代测序技术在此领域的应用,使更多的成年、晚期、耐药的Burkitt淋巴瘤患者从中获益。
