Characterization of hepatitis B virus X gene quasispecies complexity in mono-infection and hepatitis delta virus superinfection
2019-04-20CristinaGodoyDavidTaberneroSaraSopenaJosepGregoriMariaFrancescaCorteseCarolinaGonzlezRosarioCasillasMaralYllAriadnaRandoRosapezMartnezJosepQuerGloriaGonzlezAseguinolazaRafaelEstebanMarRiveiroBarcielaMariaButiFrancisco
Cristina Godoy, David Tabernero, Sara Sopena, Josep Gregori, Maria Francesca Cortese, Carolina González,Rosario Casillas, Marçal Yll, Ariadna Rando, Rosa López-Martínez, Josep Quer, Gloria González-Aseguinolaza,Rafael Esteban, Mar Riveiro-Barciela, Maria Buti, Francisco Rodríguez-Frías
Abstract BACKGROUND Hepatitis delta virus (HDV) seems to strongly suppress hepatitis B virus (HBV)replication, although little is known about the mechanism of this interaction. Both these viruses show a dynamic distribution of mutants, resulting in viral quasispecies. Next-generation sequencing is a viable approach for analyzing the composition of these mutant spectra. As the regulatory hepatitis B X protein(HBx) is essential for HBV replication, determination of HBV X gene (HBX)quasispecies complexity in HBV/HDV infection compared to HBV monoinfection may provide information on the interactions between these two viruses.AIM To compare HBV quasispecies complexity in the HBX 5' region between chronic hepatitis delta (CHD) and chronic HBV mono-infected patients.METHODS Twenty-four untreated patients were included: 7/24 (29.2%) with HBeAgnegative chronic HBV infection (CI, previously termed inactive carriers), 8/24(33.3%) with HBeAg-negative chronic hepatitis B (CHB) and 9/24 (37.5%) with CHD. A serum sample from each patient was first tested for HBV DNA levels.The HBX 5' region [nucleotides (nt) 1255-1611] was then PCR-amplified for subsequent next-generation sequencing (MiSeq, Illumina, United States). HBV quasispecies complexity in the region analyzed was evaluated using incidencebased indices (number of haplotypes and number of mutations), abundancebased indices (Hill numbers of order 1 and 2), and functional indices (mutation frequency and nucleotide diversity). We also evaluated the pattern of nucleotide changes to investigate which of them could be the cause of the quasispecies complexity.RESULTS CHB patients showed higher median HBV-DNA levels [5.4 logIU/mL,interquartile range (IQR) 3.5-7.9] than CHD (3.4 logIU/mL, IQR 3-7.6) (P = n.s.)or CI (3.2 logIU/mL, IQR 2.3-3.5) (P < 0.01) patients. The incidence and abundance indices indicated that HBV quasispecies complexity was significantly greater in CI than CHB. A similar trend was observed in CHD patients, although only Hill numbers of order 2 showed statistically significant differences (CHB 2.81, IQR 1.11-4.57 vs CHD 8.87, 6.56-11.18, P = 0.038). There were no significant differences in the functional indices, but CI and CHD patients also showed a trend towards greater complexity than CHB. No differences were found for any HBV quasispecies complexity indices between CHD and CI patients. G-to-A and C-to-T nucleotide changes, characteristic of APOBEC3G, were higher in CHD and CI than in CHB in genotype A haplotypes, but not in genotype D. The proportion of nt G-to-A vs A-to-G changes and C-to-T vs T-to-C changes in genotype A and D haplotypes in CHD patients showed no significant differences. In CHB and CI the results of these comparisons were dependent on HBV genotype.CONCLUSION The lower-replication CHD and CI groups show a trend to higher quasispecies complexity than the higher-replication CHB group. The mechanisms associated with this greater complexity require elucidation.
Key words: Hepatitis B virus; Hepatitis delta virus; Hepatitis B X gene; Next-generation sequencing; Viral quasispecies; Hepatitis B virus-hepatitis delta virus interaction
INTRODUCTION
An estimated 257 million people worldwide are chronically infected with hepatitis B virus (HBV), and 15 to 20 million of them are also infected with hepatitis delta virus(HDV)[1]. HDV is a defective RNA virus that requires the helper function of HBV surface antigen (HBsAg) to achieve transmission[2]. HDV infection can occur as an acute coinfection (simultaneous HBV/HDV infection) or as a superinfection in individuals already chronically infected with HBV[3]. Acute HBV/HDV coinfection is usually self-limited and shows a course similar to that of acute HBV mono-infection,with clearance rates of both agents greater than 95% in immunocompetent adults.Nonetheless, it can also cause severe acute hepatitis with a high-risk of developing a fulminate course[4]. In superinfections, HDV progresses to chronicity in > 80% of cases[4]. Chronic HDV infection leads to more severe liver disease than chronic HBV mono-infection. In fact, it is the most severe form of viral hepatitis in humans, with accelerated progression of fibrosis, a slightly increased risk of hepatocellular carcinoma (HCC), and early decompensation in the setting of established cirrhosis[2].
HBV is an enveloped DNA virus consisting of a 3.2-kb partially double-stranded genome that replicates via an RNA intermediate[5]and encodes 7 proteins: PreCore,core, pol, X (HBx), and the three envelope proteins, L, M, and S. The HDV genome is comprised of a 1.7-kb single-stranded circular RNA of negative polarity, the smallest among known mammalian viruses[6]. As HDV does not encode an RNA-dependent RNA polymerase, its replication relies on the host cell's RNA polymerase II (RNA pol II)[7]. The HDV genome contains a single functional open reading frame (ORF)encoding hepatitis delta antigen (HDAg). Through the action of RNA-specific adenosine deaminase 1 (ADAR 1) editing during replication, HDV manages to produce two HDAg isoforms from this single ORF: The small (S-HDAg) and large (LHDAg) delta antigens. As compared to S-HDAg, L-HDAg contains 19 additional amino acids at the C terminus[7]. Remarkably, the two HDAg isoforms have different functions: S-HDAg binds to HDV RNA and promotes HDV replication, whereas LHDAg inhibits replication and is known to be involved in HDV packaging by direct binding to HBsAg[8]. As the two viruses use the same envelope proteins, HDV and HBV share common attachment and entry steps. Direct contact between HBsAg and HDAg for HDV virion envelopment is considered the main interaction[9], but the two viruses must also interact with each other at different stages of their replication cycles.For example, previous studies have shown that HDV can strongly suppress HBV replication and become the predominant virus in HBV/HDV infection[10,11]. However,other patterns of predominance are also seen in HBV/HDV infection, related to the fluctuating patterns of HBV and HDV replication over time[12]. Currently, little is known about the specific mechanisms of this interaction. It has been suggested that the suppressive effect of HDV on HBV replication may be mediated by the interaction of HDAg with HBV enhancers[13], the L-HDAg-RNA pol II interaction[14], or by the antiviral activity of interferon-inducible MxA protein activated by L-HDAg[13].
As is the case of other RNA and DNA viruses that replicate by low-fidelity polymerases, HDV and HBV both exhibit high mutation rates, and their populations show a dynamic distribution of mutants. This characteristic results in a complex swarm of sequences that are highly similar, but not identical, known as a viral quasispecies[15,16]. Next-generation sequencing (NGS), also referred to as massive or deep sequencing, is an ideal approach to analyze the composition and complexity of the mutant spectra within a viral quasispecies[17,18]. This information can explain or predict the response of the virus to specific environmental changes. The HBV quasispecies in chronic mono-infection and the HDV quasispecies have been analyzed individually by NGS[19-23]. However, there are no studies comparing the quasispecies in HBV mono-infection and HBV/HDV infection using this technique.Characterization of the HBV population in each of these situations could provide valuable information on the interference between HBV and HDV. The pleiotropic,trans-activating HBx protein has an important role in regulating the HBV life cycle,host-virus interactions, and HBV-related HCC[24], and HDV/HBV interactions at this level may impair HBV replication/transcription. Therefore, the hepatitis B X gene(HBX) could be an interesting target to study potential interactions between these two viruses.
The aim of this study was to evaluate and compare the complexity of the HBV quasispecies in serum samples from HBV mono-infected patients in the chronic infection phase (CI, previously termed inactive carrier), patients with chronic hepatitis B (CHB) mono-infection, and patients with chronic hepatitis delta (CHD)superinfection, using a high-throughput NGS-based approach. We focused our study on the 5' end of the HBX coding region and its upstream non-coding region[nucleotides (nt) 1255-1611], which was also examined in a previous study by our group in patients in different stages of chronic HBV infection, including CHB and CI[23].
MATERIALS AND METHODS
Patients and samples
Twenty-four patients with chronic HBV infection in two different clinical stages including some with HDV superinfection were recruited from the outpatient clinics of Vall d'Hebron University Hospital (Barcelona, Spain). According to the guidelines of the European Association for the Study of the Liver[25], patients were classified into three groups: Hepatitis B e-antigen (HBeAg)-negative CI (7 patients, 29.2%), HBeAgnegative CHB (8 patients, 33.3%), and CHD (9 patients, 37.5%). The study was approved by the Ethics Committee of Vall d'Hebron Research Institute and all patients provided written informed consent for participation.
One serum sample from each patient with HBV DNA ≥ 2.5 logIU/mL (sensitivity limit of the PCR to amplify the target region) was selected for the study. Exclusion criteria were positive testing for hepatitis C virus (HCV) or human immunodeficiency virus (HIV) antibodies, current antiviral therapy, or liver transplantation in the 2 years before the sample was obtained.
Serological and virological determinations
HBV serological markers (HBsAg and HBeAg) and anti-HCV antibodies were tested using commercial electrochemiluminescent immunoassays on a COBAS 8000 instrument (Roche Diagnostics, Rotkreuz, Switzerland). Anti-HDV antibodies were tested using the HDV Ab kit (Dia.Pro Diagnostics Bioprobes, Sesto San Giovanni,Italy), and anti-HIV antibodies with the Liaison XL murex HIV Ab/Ag kit (DiaSorin,Saluggia, Italy). HBV-DNA was quantified by real-time PCR with a detection limit of 10 IU/mL (COBAS 6800, Roche Diagnostics, Mannheim, Germany). HDV-RNA was quantified by an in-house method[26]using the HDV RNA international standard of the World Health Organization (1st World Health Organization International Standard for Hepatitis D Virus RNA for Nucleic Acid Amplification Techniquesbased assays; PEI code number: 7657/12), with a quantification limit of 100 IU/mL.
Amplification of HBV and HDV regions of interest by next-generation sequencing
The region of the HBX gene (nt 1255 to 1611) selected for HBV sequencing is included in the 5' end of all HBX transcripts. It encompasses a non-coding upstream region (nt 1255-1373) and the 5' end of the HBX coding region (nt 1374-1611)[23]. This latter sequence encodes the N-terminal HBx domain (HBx amino acids 1-50), which acts as negative regulator of HBx transactivation and has an essential role in multiple functions of the protein. The region selected for HDV genotyping covered a 360-bp fragment of the HDV genome, from nt positions 910 to 1270. These genome regions have both been used to respectively determine HBV and HDV genotypes in previous studies[22,23].
For each sample, total viral nucleic acid was extracted from 200 μL of serum with the High Pure Viral Nucleic Acid Kit (Roche Diagnostics, Mannheim, Germany),according to the manufacturer's instructions. Molecular amplification of HBV-DNA was performed using a 3 PCR protocol. The first-round PCR was performed using external primers (forward 5'-TGTATTCCCATCCCATCATC, and reverse 5'-AGWAGCTCCAAATTCTTTATAAGG, which cover the region from nt 599 to 1936)with the following protocol: 95 °C for 5 min followed by 35 cycles of 95 °C for 20 s, 53°C for 20 s, and 72 °C for 15 s, and finally 72 °C for 3 min. The second- and third-round PCRs were performed as previously described by our group[23]. By adding another PCR to the amplification protocol described in that previous study, we were able to increase the sensitivity limit from 3.5 logIU/mL to 2.5 logIU/mL, thereby allowing sequencing of samples from patients with low HBV replicative activity (especially CI and CHD samples). Amplification of the region selected for HDV genotyping was performed by RT-nested PCR as previously described[21,22]. After amplification of the HBV and HDV regions of interest, the final products were flanked by universal M13 sequences at both ends. In the last PCR amplification [multiplex identifier (MID)PCR], a specific pair of primers was used, consisting of an M13 universal primer and a MID or barcode sequence. Each individual patient sample required a different MID.The PCR products of this amplification, also known as amplicons, were visualized as single bands on 1.5% agarose electrophoresis gel, stained with SybrSafe DNA Gel Stain (Invitrogen, United States) with 1 × TAE running buffer. PCR products from the gel were subsequently purified using the QIAquick Gel Extraction Kit (Qiagen,Hilden, Germany). Amplicon quality was analyzed using the Agilent 2200 TapeStation System with the D1000 ScreenTape kit (Agilent Technologies,Waldbronn, Germany). Purified DNA from each sample was quantified by fluorescence using the Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies,United States), adjusted to the same concentration, and pooled. The pools, one for HBV (24 amplicons) and another for HDV (9 amplicons), were NGS-sequenced on the MiSeq Platfrom (Illumina, San Diego, United States) following the manufacturer's protocol.
Data treatment
PCR artefacts and sequencing errors can occur when using NGS. Thus, the sequences obtained (referred to as reads: Sequences obtained by NGS that do not always cover the full amplicon length) require bio-informatic processing to minimize the scoring of these errors. To this end, we developed a haplotype-centric data analysis pipeline to exclude full reads that did not meet minimum quality requirements, essentially consisting of the following steps:
Quality control of fastq files:Inspect profiles for per-site quality, read length, and general quality-related instrument parameters.
Overlapping paired reads:In paired-end experiments, use FLASh[27]to impose a minimum of 20 overlapped base pairs (bp) with a maximum of 10% mismatches(yield 60%-80% for 450 to 500-bp amplicons).
Discarding reads:Reads are discarded if more than 5% of bases are below a Phred score[28]of 30, corresponding to an estimated accuracy of 99.9% (yield 75%-85%).
Demultiplexing reads:Demultiplexing is done by identifying oligonucleotide sequences at both ends within windows of expected positions in the reads (yield 70%-85%). First, the individual MIDs (10 oligonucleotide sequences) are used to distinguish between samples from different patients/origins. Only one mismatch is allowed. Second, specific primers (20 to 30-bp oligos) are used to distinguish between different regions in the genome or different genomes, and between the two strands.Up to three mismatches are allowed. Finally, MIDs and primers are trimmed and a fasta file is obtained for each combination of MID, primer, and strand in the run,where reads are collapsed to haplotypes (unique sequences covering the full amplicon observed on the clean set of sequences) with the corresponding frequencies.
Aligning haplotypes:In each fasta file haplotypes are aligned to the wild-type reference sequence or the master sequence (most abundant haplotype in the file) and quality filter (yield > 90%). This quality filter consists in discarding haplotypes that do not cover the full amplicon and those that have more than two indeterminations, three gaps, or more than 99 differences with respect to the reference. Finally, the accepted indeterminations and gaps are repaired as per the reference sequence.
Intersecting haplotypes: For this step, haplotypes with abundance not below 0.1% in both strands are selected (yield 50%-60%), whereas those unique to one strand are discarded. The coverage of haplotypes passing the filter is taken as the sum of reads in both strands.
Final result:All haplotypes with abundances not below 0.25% are kept. The final haplotypes are called consensus haplotypes, and these are the basis for the downstream analysis in this study (final overall yield 15%-25%).
Genotyping
HBV and HDV genotypes were determined by NGS and phylogenetic analysis of the amplified genome regions of both viruses. NGS allows detection of mixtures of viral genotypes in patient samples, which could have an impact on HBV quasispecies complexity. The nt haplotypes aligned at 0.25% obtained by NGS were genotyped by distance-based discriminant analysis (DB rule)[29,30]. For this analysis, we used reference sequences of the HBV and HDV regions analyzed extracted from the fulllength genomes representative of HBV genotypes A to H and HDV genotypes 1 to 8 obtained from GenBank (Supplementary Figures 1 and 2). This analysis takes into account the inter- and intra-class variability of each genotype. Genetic distances were computed according to the Kimura-80 model[31]. UPGMA trees were designed to visualize the genetic distances between sequences.
Quasispecies complexity measures

Figure 1 Comparison between hepatitis B virus-DNA serum levels in the three groups.
Quasispecies complexity was analyzed in HBV sequences obtained by NGS (5' HBX gene, nt 1255-1611). Multiple alignment displays the entities (haplotypes,polymorphic sites, and mutations) present in the viral quasispecies. In this study, six parameters were used to describe quasispecies complexity: Number of haplotypes(nHpl) and number of mutations (nMuts) as incidence-based indices; Hill numbers of order 1 and 2 (q = 1, the exponential of Shannon's entropy and q = 2, the inverse of Simpson's index) as abundance-based indices; and the mutation frequency (Mf) and nucleotide diversity (Pi) as functional indices[18,32]. The complexity parameters are defined in Supplementary Materials.
Analysis of nucleotide substitutions
The innate immune system is suggested to contribute to HBV genetic variability through the cytidine deaminase APOBEC (apolipoprotein B mRNA editing enzyme)family[33], such as APOBEC3G (A3G) which promotes G-to-A, and in some cases, C-to-T hypermutation of HBV genomes[34,35]. To infer whether this nt substitution pattern could be associated with HBV quasispecies complexity in the groups analyzed, we used a point mutation approach to detect bias towards a specific nt change. This approach consists in assessing the nt changes in the sequence of haplotypes relative to the reference sequence of the same genotype (consensus sequence of all GenBank patterns of the same genotype used for HBV genotyping), taking into account only one nucleotide change per position, regardless of the number of haplotypes where it appears.
In addition, we compared the proportion of positions with a G-to-A nt change in any haplotype (relative to the genotype reference sequence) with the proportion of positions with an A-to-G nt change (G→A/G vs A→G/A) between CHB, CI and CHD. The same comparison was done for C-to-T changes: (C→T/C vs T→C/T).
Statistical analysis
Statistics were carried out using GraphPad Prism version 7.0 (GraphPad software, La Jolla, United States). All parameters are expressed as the median value and interquartile range (IQR). For qualitative variables the chi-square test was performed.The Kruskal Wallis and Dunn test (post hoc) were used for multiple comparisons of independent samples. To compare the proportions of nt changes, a 2-sample test for equality of proportions with continuity correction was performed. The Mann-Whitney test was used for the two-group comparisons (HBeAg+ vs HBeAg- in CHD, and cirrhotic vs non-cirrhotic patients). P values < 0.05 were considered significant. The bioinformatics and biostatistics methods used in this study were reviewed by Dr.Josep Gregori from the Liver Disease-Viral Hepatitis Laboratory of Vall d'Hebron Hospital (Barcelona, Spain), CIBERehd research group, and Roche Diagnostics SL.
RESULTS
Patient characteristics
Clinical, virological, and serological parameters were obtained from the 24 patients.Baseline characteristics are summarized in Table 1. Median (IQR) age was 37 (30-45 years), 58% were men, 71% were Caucasians, and the remainder were of Sub-Saharan origin. Regarding laboratory characteristics, 79% were negative for HBeAg and median (IQR) alanine aminotransferase levels were 43 IU/L (31.75-118.5). HBV DNA levels were lower in the CI [3.2 logIU/mL (2.3-3.5)] and CHD [3.4 logIU/mL (3-7.6)]groups than in CHB [5.4 logIU/mL (3.5-7.9)] (P < 0.01 and n.s., respectively) (Figure 1). Liver histology was available in all cases: 2/9 patients with CHD (1 of whom additionally had HCC), and 3/8 patients with CHB had liver cirrhosis.
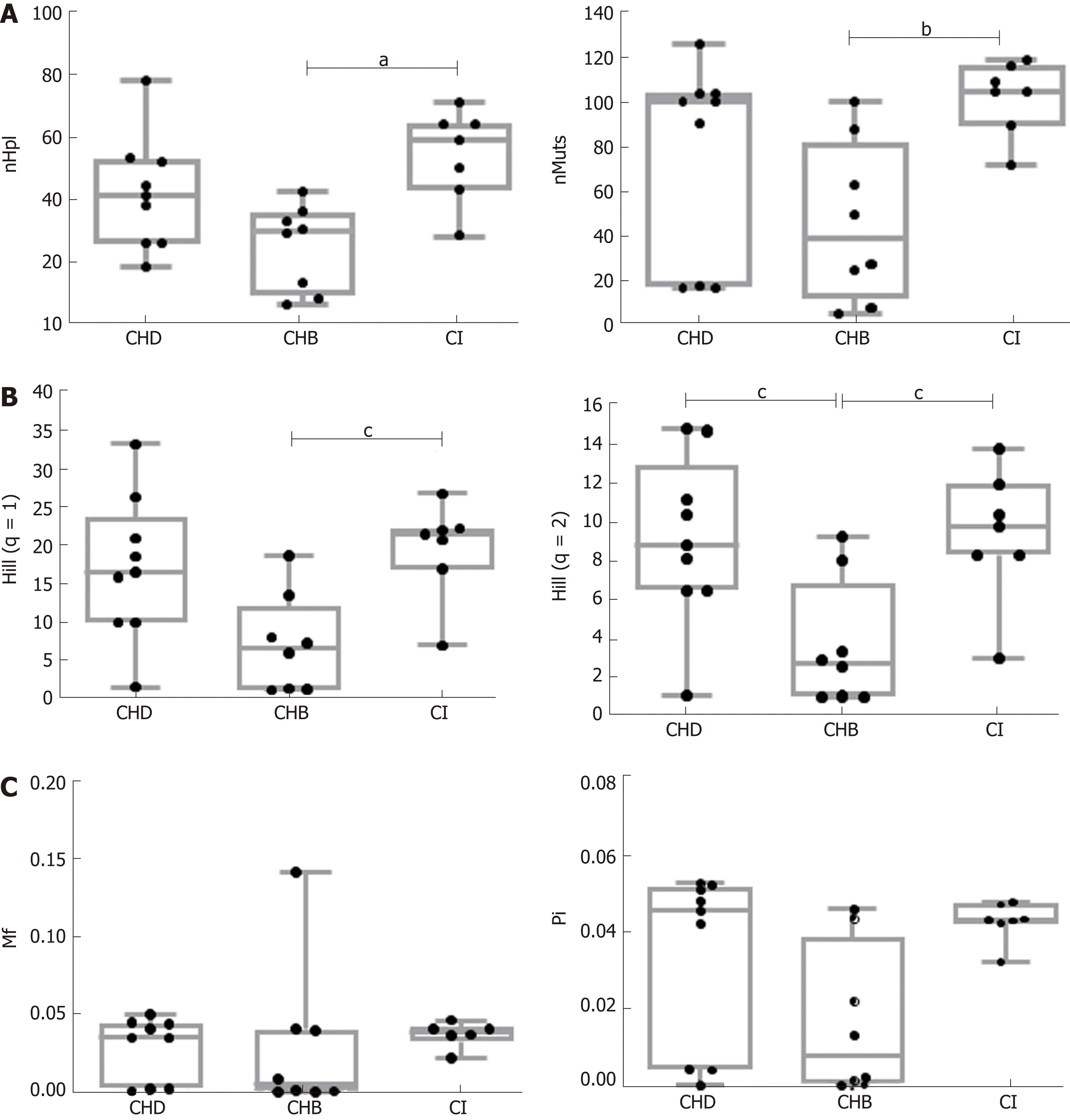
Figure 2 Comparison of the indices of hepatitis B virus quasispecies complexity between the three groups.
Analysis of NGS sequences obtained and genotyping results
After applying the quality filters, 791036 sequences from the HBV target region were obtained from the 24 serum samples, yielding a median (IQR) of 26459 (17649-44852)sequences per patient. Regarding HDV, 287541 sequences were obtained from the 9 samples, yielding a median (IQR) of 17609 (14475-22677) sequences per patient. HBV genotyping showed that in the region analyzed (nt 1255 to 1611), 15/24 patients(62.5%) had a complex mixture of genotypic variants, mainly A/D/C. None of the patients included showed genotype B, G, or H haplotypes. With regard to the HDV region analyzed (nt 910 to 1270), 7/9 (78%) CHD patients were classified as genotype HDV-1, whereas 2/9 (11%) were HDV-2 and HDV-6, respectively.
Characterization of HBV quasispecies complexity
To analyze HBV quasispecies complexity data from samples of notably different coverage, samples were made comparable by down-sampling and fringe trimming to a common coverage of 6000 reads, retaining haplotypes at a frequency above 0.2%with 95%CI. Quasispecies complexity in the HBV target region was evaluated using six parameters. Of note, all CHB and CI patients were HBeAg-negative, whereas 5/9 CHD patients were HBeAg-positive. The complexity of the viral population in the preCore/Core region of the viral genome has been reported to differ between HBeAgpositive and -negative HBV mono-infected patients[36]. To determine whether HBeAgstatus had an effect on quasispecies complexity in the HBX 5' region in CHD patients,the six parameters used to assess this factor were compared between HBeAg-negative and -positive CHD patients, but no statistically significant differences were found(Table 2). Thus, the patients' HBeAg status was not considered to be a potential interfering factor. In addition, we compared these complexity indices between 5 patients with liver cirrhosis, including 2 CHD and 3 CHB, and 19 patients without progression to severity. No significant differences were found (Table 3).
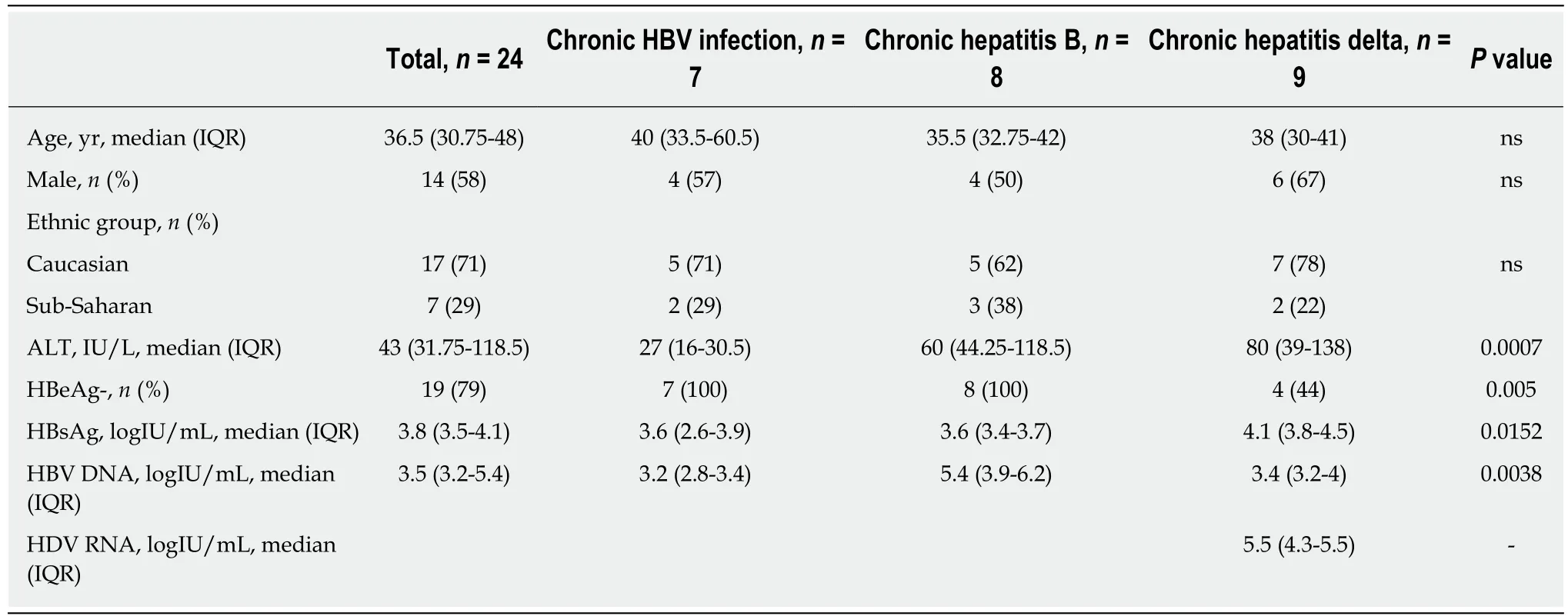
Table 1 Baseline characteristics according to the clinical stage of hepatitis B virus or hepatitis B virus/hepatitis delta virus infection
Comparison between HBV mono-infection (CHB and CI) vs. CHD (HBeAgnegative and HBeAg-positive) in two-group tests showed no significant differences in the incidence, abundance, or functional indices related to quasispecies complexity.However, significant differences were found between CHB and CI in incidence and abundance-that is, median nHpl (IQR), CHB 31 (12-75, 34.75) vs CI 60 (47.5-65) (P =0.011), nMuts, 39 (20.75-69.25) vs 105 (97.5-112.5) (P < 0.01), and Hill numbers of order 1, 6.70 (1.37-9.43) vs 21.54 (18.99-22.14) (P = 0.012), and 2, 2.81 (1.11-4.57) vs 9.86 (8.37-11.22) (P = 0.027)-with the CI quasispecies showing greater complexity than that of CHB (Figure 2). No statistically significant differences were observed for the functional indices (Mf and Pi), which are sensitive to the number of differences between the different haplotypes, although the results showed a trend towards greater complexity in CI and CHD than in CHB patients (Figure 2C). Regarding the effect of HDV on the HBV quasispecies, the most interesting finding was that the HBV viral populations in CHD and CI showed a similar trend, with greater complexity(higher incidence, abundance and functional values) than that of CHB patients,although only Hill numbers of order 2 showed a statistically significant difference,CHB 2.81 (1.11-4.57) vs CHD 8.87 (6.56-11.18) (P = 0.038) (Figure 2). There were no significant differences in any HBV complexity indices between CHD and CI patients.
Types of nucleotide changes in HBV sequences
All types of nt changes were computed in the three patient groups. As the patterns of nt changes varied between the different genotypes (data not shown), we only took into account genotype A (Figure 3A) and D (Figure 3B) haplotypes, which were the most abundant in our samples [genotype A 511/1065 haplotypes (47.98%) and genotype D 264/1065 haplotypes (24.79%)], for the comparisons. In this analysis we found that the pattern of nt changes in genotype A haplotypes (Figure 3A) differed from those in genotype D (Figure 3B). The nt changes G-to-A and C-to-T, which are characteristic of the modifications introduced by the A3G enzyme, were higher in CHD and CI than in CHB patients in genotype A haplotypes (Figure 3A), but not in genotype D (Figure 3B). We then compared the proportion of G-to-A vs A-to-G nt changes and C-to-T vs T-to-C in genotype A and D haplotypes by group to investigate bias in the nt change pattern, which could be associated with the effect of A3G. There were no significant differences in these changes in either genotype A or genotype D in CHD patients. The results in CHB and CI were dependent on HBV genotype. In genotype A haplotypes, the proportion of C-to-T nt changes was higher than T-to-C in the CI group (0.22 vs 0.07 respectively, P < 0.01), whereas in genotype D the proportion of G-to-A nt changes was higher than A-to-G in the CHB group (0.20 vs0.04 respectively, P = 0.025).

Table 2 Comparison of hepatitis B virus quasispecies complexity indices between hepatitis B e-antigen-negative and hepatitis B eantigen-positive chronic hepatitis D patients
DISCUSSION
Clinical and experimental data support the existence of interference between HDV and HBV. Although several hypothesis have been raised to define the interactions between these viruses[37], the molecular mechanisms through which HDV affects HBV remain elusive. In the clinical setting, most HBV/HDV co-infected patients show a pattern of HDV dominance, with a significant decrease in HBV-DNA viral load when compared to that of mono-infected patients[12,38,39]. In accordance with these data, our results showed lower HBV-DNA levels in HDV-infected patients, with values similar to those in the chronic HBV infection group.
The viral population analyses carried out found no significant differences in the complexity of the HBV quasispecies between the total of patients with HBV monoinfection and patients with CHD. These results would seem to suggest that HBV quasispecies complexity is unaffected by the presence of HDV. However, when the clinical phases of the disease were taken into account, HBV quasispecies complexity was found to be greater (significant in incidence and abundance-based indices and non-significant in functional indices) in CI than in CHB patients. Surprisingly, the viral population was more complex in the group with lower replication. This may indicate that the higher incidence of mutations in the 5' of HBX, distributed in different haplotypes in CI patients could cause HBV replication to be closer to the quasispecies error threshold, that is, the point beyond which the mutation rate is so high that the genetic information carried by the replicating genome is lost[40]. In this sense, it should be taken into account that HBx is essential for HBV replication. For this reason, it seems logical to associate HBX variability with HBx functionality, which would affect HBV replication and result in the low HBV replication levels observed in the CI stage of HBV infection.
As to the interaction between HDV and HBV, it seems that CHD drives the HBV quasispecies to a situation similar to that observed in CI patients: Lower replication level and higher HBX quasispecies complexity than CHB patients. Two hypotheses could explain the mechanism by which HDV enhances HBV quasispecies complexity.The first is activation of the host innate immune response under the effect of HDV stimulation[39,41,42]. A3G activity, which provides broad innate immunity[34,43], could therefore be responsible for the hyper-mutation of HBV genomes. To investigate this possibility, we analyzed nucleotide changes in the CHB, CI, and CHD groups to determine whether there was some bias in favor of those produced by A3G. Although HDV activates the immune system, we did not find a hyper-mutation pattern associated with A3G. Nonetheless, this hypothesis should be more extensively analyzed in further, more specific studies, and it could be extended to other innate immunity enzymes.
The second hypothesis postulates a possible interaction between HDAg and RNA pol II, which could affect the replicative capacity and functionality of this enzyme. As Yamaguchi et al[14]reported, HDAg not only increases the elongation rate, it also reduces transcriptional fidelity by interacting with and loosening the RNA pol II clamp. This would increase the error rate, introduce a larger number of mutations,and give rise to a more complex quasispecies. This mechanism has been suggested to explain the extremely high mutation rate that occurs in HDV replication[22,44]. In addition to HDV, this loss of fidelity would also affect HBV transcription. It isimportant to keep in mind that HBV cccDNA is the template for transcription of all viral mRNA including pregenomic RNA, essential for progeny production, and that pregenomic RNA transcription from both cccDNA and integrated HBV DNA is mediated by the host RNA pol II[45,46]. This effect of HDV on RNA pol II could also affect cellular mRNAs, thereby worsening cell homeostasis, and this could be linked to the poorer prognosis of HDV hepatitis when compared to the other viral hepatitis.To test this possibility, NGS studies investigating the complexity of cellular mRNAs in CHD are needed.
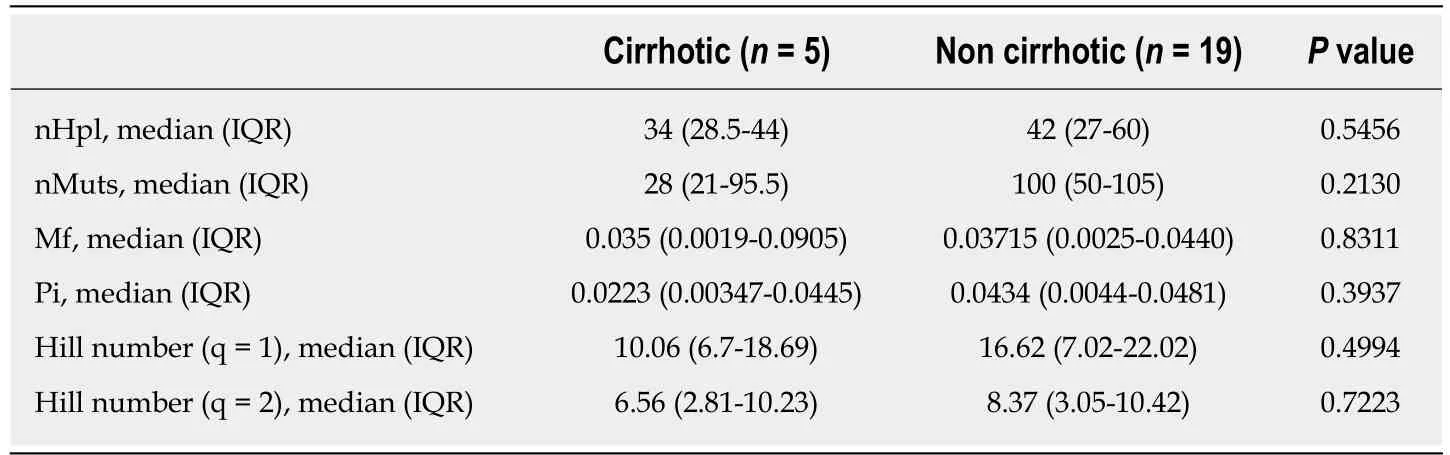
Table 3 Comparison of hepatitis B virus quasispecies complexity indices between patients with and without liver cirrhosis
Thus, hyper-mutation due to the innate immune system and loss of RNA pol II fidelity by the effect of HDV could have an impact on HBV DNA synthesis, which,along with the error rate of the HBV polymerase itself, could give rise to a situation similar to that seen in CI patients, driving the HBV quasispecies closer to its error threshold. However, we would expect to find significant differences between CHD and CHB in other complexity indices in addition to Hill numbers (q = 2) in this scenario. Hence, larger samples taken at different time points and other regions of the genome should be analyzed in future studies to confirm these results.
In this line, 2 recent studies[47,48]investigated HBV sequence variation in the viral genome surface (S) ORF (encoding HBsAg) in large patient cohorts. The authors compared consensus sequences obtained from HBV/HDV infected vs HBV monoinfected patients. Both studies concluded that HDV can exert selective pressure over some positions of the S ORF, constraining HBV evolution. In the light of these findings, it would be interesting to assess the effect of HDV on the HBV quasispecies in the S ORF and compare it with the effect in other regions of the viral genome, such as that analyzed in the present study. These efforts illustrate the relevance of studies investigating the HBV quasispecies in HDV superinfected or coinfected patients to deepen current knowledge on the interference between HDV and HBV. In addition,cellular and animal models of HBV/HDV infection[42,49]enable in vitro and in vivo functional studies to test whether the presence of HDV has an effect on HBV replication and genetic diversity.
Of note, the patients included in this study had to have HBV replication at high enough levels for amplification by our PCR protocol. This obliged us to include HBeAg-positive CHD patients (5/9), whose numbers are limited in HBV/HDV infection. We believed it was necessary to determine whether HBeAg status may have had an effect on quasispecies complexity, as we found a more complex viral population in the preCore/Core region of the HBV genome in HBeAg-negative than HBeAg-positive CHB patients in a previous analysis[36]. However, the present study,focusing on the 5' region of HBX (nt 1255-1611), showed that HBV quasispecies complexity was similar in HBeAg-positive and -negative CHD. These differences may be related to the regions studied: the 5' region of HBX does not have a direct relationship with HBeAg status; hence, it would not have a significant influence on HBV quasispecies complexity in this region. Thus, as HBeAg status did not seem to significantly affect HBV quasispecies complexity in the 5' HBX region, we were able to compare all CHD patients (both HBeAg-positive and HBeAg-negative) with CHB and CI patients. Another factor that could be related to HBV quasispecies complexity is liver disease progression to cirrhosis or HCC, which had occurred in 5 patients(both CHB and CHD) included in this study. Comparison of quasispecies complexity between these patients and the 19 who did not progress to severity showed no statistically significant differences. Nonetheless, although HBeAg status and more severe disease stage did not affect HBV quasispecies complexity in our sample,analysis of larger patient groups is needed to define the actual role of these virological and clinical factors.
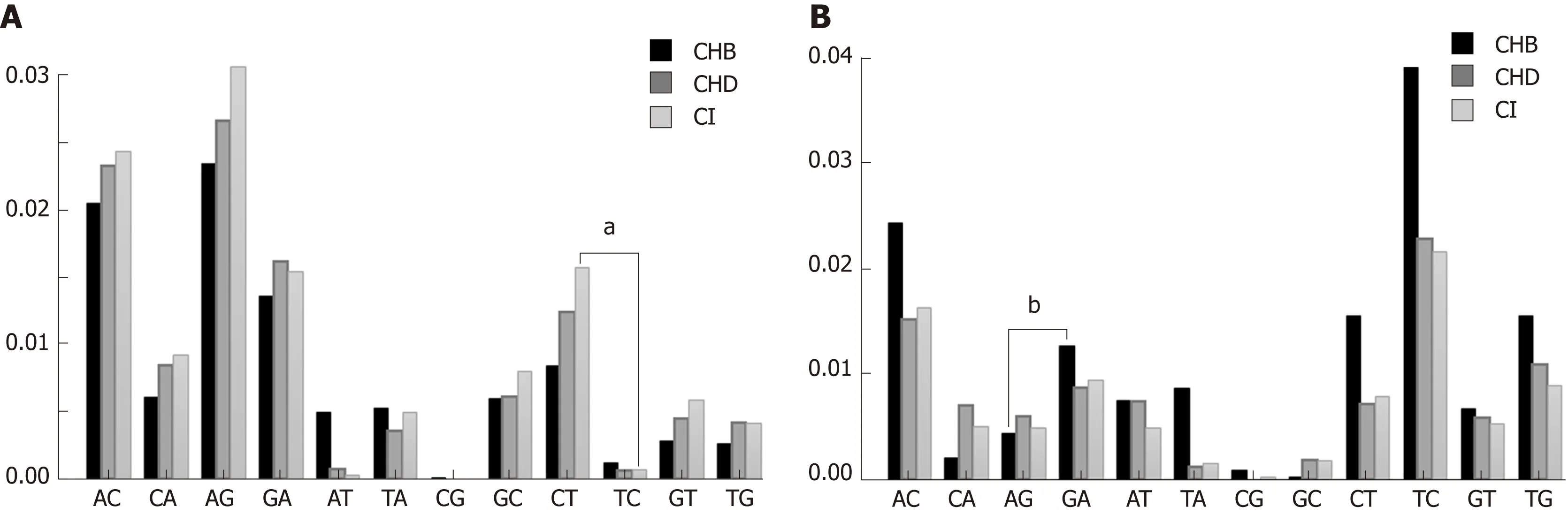
Figure 3 Nucleotide change patterns.
In summary, this study provides the first data on the influence of HDV on HBV genetic diversity in the HBX gene, obtained using NGS. Our results showed that in HBV stages with lower replication (CHD and CI), the HBV quasispecies in the 5'end of HBX exhibited a trend toward higher complexity than in CHB. This was mainly evident in terms of incidence and abundance, that is, a higher incidence of mutations,distributed in different haplotypes. The mechanisms associated with this greater complexity are unknown, but two hypotheses could explain them: involvement of the innate immune response or HDAg interaction with RNA pol II, which should be explored in greater depth.
ACKNOWLEDGMENTS
The authors thank Celine Cavallo for English language support and helpful editing suggestions.
ARTICLE HIGHLIGHTS
Research background
Hepatitis delta virus (HDV) causes the most severe form of chronic viral hepatitis in persons simultaneously infected with hepatitis B virus (HBV). In longitudinal clinical studies, HDV infection has been associated with a considerable temporary or permanent reduction in HBV viral load, whereas HBV surface antigen levels are usually high. Thus, beyond the interaction with HBV envelope proteins, there are other mechanisms by which HDV inhibits HBV-DNA replication.
Research motivation
To date, little information has emerged on the interaction between HDV and HBV. In this study,we investigated whether HDV can affect the complexity of the HBV quasispecies, and proposed possible mechanisms by which it may do so, to further characterize the interaction between these two viruses.
Research objectives
Considering the essential role of the HBV X protein (HBx) on viral replication, the aim of this study was to analyze the 5' end of the hepatitis B X gene (HBX) coding region and its upstream non-coding region (nt 1255-1611) by next-generation sequencing (NGS) to evaluate HBV quasispecies complexity between chronic hepatitis delta (CHD)-infected patients and chronic HBV mono-infected patients [HBV chronic infection (CI) and chronic hepatitis B (CHB)].
Research methods
The HBX 5' end region, nucleotide (nt) 1255-1611, was PCR-amplified for subsequent NGS(MiSeq, Illumina, United States) in 7 CI, 8 CHB, and 9 CHD patients. HBV quasispecies complexity in the region analyzed was evaluated using incidence-based indices [number of haplotypes (nHpl) and number of mutations (nMuts)], abundance-based indices (Hill numbers of order, q = 1 and q = 2) and functional indices [mutation frequency (Mf) and nt diversity (Pi)].The pattern of nt changes was evaluated to investigate the cause of HBV quasispecies complexity.
Research results
HBV quasispecies complexity was significantly higher in the CI group than in CHB for abundance (Hill numbers q = 1 and q = 2) and incidence (nHpl and nMuts). In CHD, the HBV quasispecies showed a trend towards higher complexity similar to that of CI patients. No significant differences were observed in Mf or Pi between the groups, although CI and CHD showed a trend towards greater quasispecies complexity than CHB patients. The proportion of G-to-A vs. A-to-G and C-to-T vs. T-to-C nt changes in genotype A and D haplotypes by group did not provide conclusive evidence of a hyper-mutation pattern associated with the innate immune system enzyme APOBEC3G.
Research conclusions
The HBV quasispecies showed a trend to higher complexity in groups with lower viral replication (CHD and CI) than in the higher-replicating CHB patients. This could indicate that HDV has an effect on the 5' HBX sequence, increasing HBV quasispecies complexity. Two different mechanisms are proposed to explain how HDV can change the HBV quasispecies:hypermutation by activation of the innate system through HDV stimulation or loss of RNA pol II fidelity due to its interaction with hepatitis delta antigen. Further studies are needed to determine the clinical impact of the increased HBV quasispecies complexity in CHD patients,which may be of help to devise new therapy strategies.
Research perspectives
CHD drives the HBV quasispecies to a situation similar to that found in HBV CI: Lower replication level and higher HBX quasispecies complexity than in CHB patients. Further studies are needed to characterize the mechanisms by which HDV acts on the HBV quasispecies, which may include the innate immune system or RNA pol II fidelity.
杂志排行

World Journal of Gastroenterology的其它文章
- Comprehensive and innovative techniques for laparoscopic choledocholithotomy: A surgical guide to successfully accomplish this advanced manipulation
- Hepatocellular carcinoma surveillance: An evidence-based approach
- Cellular therapy: A promising tool in the future of colorectal surgery
- Plasma microRNAs as potential new biomarkers for early detection of early gastric cancer
- Comparison of Hemospray® and Endoclot™ for the treatment of gastrointestinal bleeding
- Performance of tacrolimus in hospitalized patients with steroidrefractory acute severe ulcerative colitis