液相色谱—质谱法测定血药浓度试验中提高灵敏度的方法概述
2019-04-02马智宇
摘 要 血浆中药物(或内源性物质)浓度检测是药代动力学的基础,也是药物临床前和一期临床研究重要组成部分,还是一些疾病和胎儿早期筛查的重要指标。目前血浆中药物浓度检测广泛采用液相色谱-质谱法(LC-MS/MS)。而LCMS/MS测定过程中,常遇到药物的检测灵敏度不够的问题。检测灵敏度不够主要是由于药物浓度过低或药物结构质谱响应低造成的。本文概述了LC-MS/MS法血药浓度检测中提高灵敏度的主要途径,即根据药物结构,从质谱方法、色谱方法、样品制备方法和避免低浓度样品污染四个方面来联合提高药物检测灵敏度的解决方案,从而为解决血浆中药物检测灵敏度不够的困难提供帮助。
关键词 LC-MS/MS 低灵敏度 超低浓度
中图分类号:O657; R969.1 文献标志码:A 文章编号:1006-1533(2019)05-0074-05
An over view of methods for improving sensitivity in the determination of plasma drug concentration by liquid chromatography-mass spectrometry
MA Zhiyu1,2*(1. Shanghai Pharmaceuticals Holding Co., Ltd., Shanghai 201203, China; 2. Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China)
ABSTRACT The determination of drug (or endogenous substance) concentration in plasma is the basis of pharmacokinetics, an important component of preclinical and phase I clinical studies and also an important indicator of some diseases and early fetal screening. At present, the determination of drug concentration in plasma is mainly performed by liquid chromatography-mass spectrometry (LC-MS/MS). The most difficult problem often encountered in the LC-MS/MS analysis is the one of insufficient sensitivity of drug determination, which is mainly due to the low drug concentration or the low response of the drug structure to mass spectrometry. This paper outlines the main ways to improve the sensitivity of LC-MS/MS for the determination of plasma drug concentration, and the solutions to improve drug determination sensitivity are described based on the drug structure from four aspects such as the mass spectrometry method, chromatographic method, sample preparation method and avoidance of low concentration sample contamination so as to provide helps for solving the difficulty of insufficient sensitivity of drug determination in plasma.
KEY WORDS LC-MS/MS; low sensitivity; ultra-low concentration
測定血药浓度是研究药代动力学的基础,而动物和人的药动学研究是药物临床前研究和一期临床安全性评价重要组成部分,是新药上市之前研发必由之路,同时还是疾病和胎儿早期筛查的重要途径。由于血浆中含有血浆蛋白、盐离子、水分和待测药物等复杂组分,且其中药物浓度相对较低,而液相色谱-质谱法(LC-MS/ MS)集分离和检测技术为一体,能把药物从体内大多数复杂物中分离出来进行测定,故成为目前测定血浆中药物浓度的主要方法[1-6]。
药物血浆浓度检测中,由于检测灵敏度不够而不能满足测定要求是最常见的问题[7-8]。其原因一方面是待测药物血浆中浓度过低,另一方面是药物本身结构响应低或质谱离子化不稳定,比如某些类固醇类药物不含质谱响应基团或某些药物(如鱼腥草素)质谱中裂解[3-6,9]。以上的两种问题都需要提高仪器对药物检测灵敏度来满足测定要求。本文将对LC-MS/MS法测定血浆中药物如何提高检测灵敏度的方法进行概述,为该类药物的血浆浓度测定提供参考,从而为药物研发提供帮助。在建立液相色谱-质谱仪(LC-MS/MS)测定血浆中药物方法时,一般从三方面进行优化:质谱方法、色谱方法和样品制备方法。本文将分别从这三个方法来概述建立此类方法的具体注意事项。
1 质谱方法
待测药物在质谱仪测定时的响应往往和药物本身的化学结构、质谱仪所用离子源、质谱仪相关参数和待测药物形成的加和离子类型或裂解离子等因素有关[7-12]。
1.1 离子源类型
目前的液相色谱-质谱仪(LC-MS/MS)通常有三种离子源:ESI源(电喷雾离子化源)、APCI源(大气压化学电离源)和APPI源(大气压光电离源)。而在药学领域,主要应用ESI源和APCI源。它们使药物离子化的方式不同,ESI源是使药物在液体状态下带电,APCI源是使药物在气体状态下离子化。故而ESI源适合于大多数有一定极性的药物,而APCI源则适合极性较弱的气体状态不容易裂解的药物。当药物化学结构极性相对较大时,往往优先选用ESI源;但药物极性较小时,APCI源则因为测定时仪器响应稳定而更加有优势[7-8]。
1.2 质谱相关参数
质谱相关参数包括三种类型:质谱硬件状态、质谱系统参数和药物自身质谱参数[7-8,11-14]。
质谱硬件状态主要是指喷雾针相对位置,在调试时,可通过喷雾针的微调旋钮来对喷雾进行调节,以获得最佳的质谱响应值。喷雾针相对位置往往和流动相流速和组成相关。
质谱系统参数包括喷雾电压、气化温度、雾化器压力和温度、反吹气压力和温度等。这些参数的调试要结合试验的色谱方法来进行,例如流动相流速大或水相占比高,气化温度、雾化器压力和温度等参数就要相应的提高,以保证流动相被完全喷散并雾化,从而增大仪器响应值。而反吹气则是在仪器背景噪音较高,样品基质相对较脏时使用,可降低背景噪音,从而提高相对灵敏度。在进行多个药物同时测定时,以上参数应以浓度最低或检测灵敏度最低药物为基准。
药物自身质谱参数主要包括去簇电压和碰撞能量。这两个参数仅与药物本身结构和碎片离子结构有关,故在优化完之后,质谱色谱系统方法再改变对此两个参数影响都不大。此外,由于大多数串联质谱都不是高分辨质谱,故而可通过调整分子量后面小数点来选择母离子和子离子,从而优化质谱响应值[10]。
1.3 加和离子和裂解离子
大部分药物在质谱中都能得到准分子离子峰,即[M+H]+峰。但也有不少药物得不到[M+H]+峰或者[M+H]+峰相对于其他加和峰响应值小很多,这时就需要选择加和离子峰作为其母离子来进行检测,以提高响应值。常见的加和峰可分为正电模式,如[M+NH4]+,[M+Na]+,[M+Li]+,[M+K]+和负电模式,如[M+Cl]?,[M+HCOO]?,[M+CH3COO]?。有文献报道选择[M+Li]+对紫杉醇类药物进行测定,相对于[M+H]+峰有更高的质谱响应值,相对于[M+Na]+峰有更稳定的质谱响应值。这是由于Li+是定量加入流动相中的,相对于[M+Na]+来说含量更稳定,而紫杉醇类药物加和峰往往比[M+H]+峰响应值要大[7,11]。
有些药物在质谱中容易得到其裂解离子峰,而得不到母离子峰或母离子较小,比如鱼腥草素和雄酮。可选择其确定的脱水裂解峰作为母离子进行定量测定[9,12]。
2 色谱方法
待测药物色谱方法的选择通常要根据药物自身的极性、pKa、结构中的基团等来进行优化。从药物自身的结构出发,我们在色谱方法建立上,主要考察流动相中有机相-水相比例、pH、缓冲盐种类和浓度、其他添加物和流动相流速及梯度[7-8,15-17]。
2.1 有机相-水相比例
在用LC-MS/MS法进行血浆药物浓度测试,通常会让药物在3倍死时间之后出峰,避开之前血浆中大极性物质基质干扰[13-14]。待测药物出峰之后用流速梯度或组分梯度进行冲洗,减少药物出峰之后的基质干扰。而调节药物在色谱柱上面的保留,就是靠调节流动相比例来进行的,药物合适的保留能尽可能减少前后基质的干扰,这样才能保证相对高且稳定的响应值。
不同的流动相组成也能影响药物在质谱上的响应值,通常情况下响应值排行:高比例有机相中>纯有机相中>高比例水相中>纯水相中。此外,有的药物在一种有机相中响应远远小于另一种有机相。通常情况下,正电药物在甲醇有机相体系中响应大于在乙腈体系中,负电药物在乙腈体系中响应大于甲醇体系[7,15-16]。
在选择流动相比例时,要同时考虑质谱响应和色谱分离两方法因素再做决定。
2.2 流动相pH
不同药物的pKa不同,故在不同pH流动相中,其分子态-离子态比率不同,最终将影响药物的离子化状态,从而对质谱响应值有影响。通常正电药物在酸性流动相下响应值较高,而负电药物在碱性流动相下响应值较高。然而流动相pH也会对药物的色谱行为产生影响,例如正电药物虽然在酸性流动相下响应值较高,但也容易形成拖尾峰或弱保留[7,15-16]。
此外,还应注意非挥发型酸碱添加剂会对质谱响应造成抑制,不同类型色谱柱对流动相pH的耐受范围也不同。因此,在选择流动相pH时应同时考虑多方面因素。
2.3 盐离子
流动相中适量的盐离子有助于药物的电离,相对于不含盐离子的流动相,药物的质谱响应值会更大。此外,流动相中盐离子的存在,可以使流动相体系形成一个缓冲盐体系,使其在加入酸或碱添加剂时,整个體系的pH相对稳定。提高并且稳定了药物的离子化效率,从而提高并且稳定了质谱的响应值[15-16]。但盐离子的浓度并非越高越好,因为浓度过高会抑制质谱响应值,也会在有机相比例增加时析出盐离子堵塞色谱流路。
和酸碱添加剂一样,添加的盐离子必须是挥发型的,因为非挥发性盐离子(如磷酸盐)会对质谱造成损害。
2.4 流速
为减少待测药物出峰前后的干扰和节省分析时间,流速梯度很常用。不同流速进入质谱对响应有明显影响,ESI源通常接受0.3~0.6 ml/min流速,APCI源通常接受0.5~0.8 ml/min流速。因此,应把待测药物出峰时间段的流速调至合适流速,而前后冲洗段可用大流速冲洗,并切出离子源流路。此外,在优化质谱响应时,应在确定色谱方法后,再对质谱系统参数进行调整,因为此类参数与流速和比例有较大相关性[8,15-17]。
3 樣品制备方法
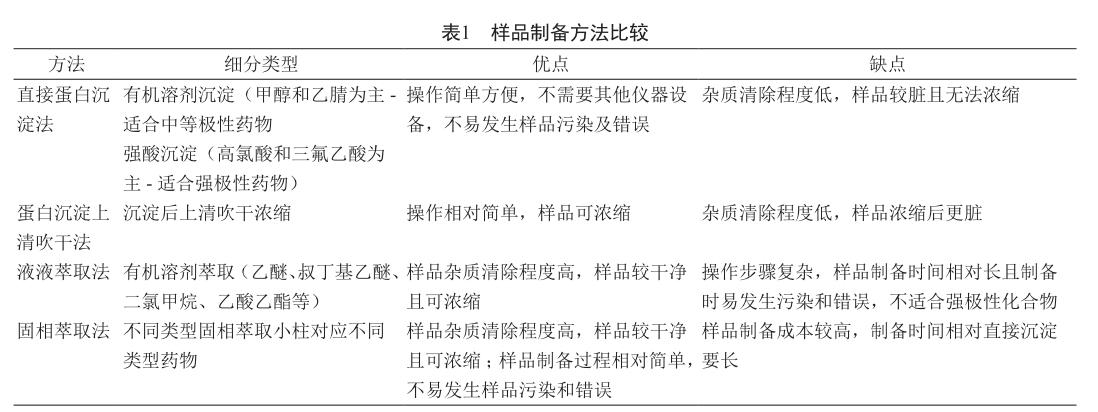
目前的血浆样品处理方法主要有直接蛋白沉淀法、蛋白沉淀上清吹干、液液萃取法和固相萃取法。每种方法各有优缺点,但目的都在于去除血浆中的内源性基质和浓缩样品等。表1中分别列举了三种方法的优缺点[18-19]。
对于提高药物检测灵敏度来说,浓缩样品和减少复杂过程中的污染问题,是两个必须兼顾考虑的问题。此外,最终进入仪器分析的样品的溶剂组成和pH也是影响质谱灵敏度的重要因素,具体要结合本文的质谱方法和色谱方法中的各方面因素综合考虑;如果待测物质谱响应非常小,也可通过衍生化法来引入响应基团,增加待测物响应[20-21]。
4 注意事项
因待测药物浓度很低,故防止样品污染问题很重要,必须从整个试验的全过程来防止样品污染。例如流动相配制过程必须远离待测药物,低浓度样品和高浓度样品全程隔开,样品检测过程Carryover效应必须完全达标,尝试不同类型的洗针方式和洗针溶液等。这个方面对测试结果准确性有直接影响,但降低本底污染也能相对提高检测灵敏度。
5 结语
随着新药研发的不断推进,低浓度药物(如某些药物活性代谢物毒性更小活性不变或更大)和低检测灵敏度药物(部分类固醇类药物)不断出现。而血浆药物浓度的监测,对于药物安全性有重大意义。因此,提高药物检测灵敏度具有重要意义。具体来说,需要从质谱方法、色谱方法和样品预处理方法三个方面协调改进,以增加检测灵敏度,为检测灵敏度较低的血浆中药物检测提供帮助,从而为该类药物的研发助力。
致谢:对中科院上海药物研究所药物代谢研究中心的全体领导、同事和学生对此综述的完稿所提供的帮助表示衷心感谢
参考文献
[1] Van den Ouweland JM, Kema IP. The role of liquid chromatography-tandem mass spectrometry in the clinical laboratory[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2012, 883-884: 18-32.
[2] Lappin G, Garner RC. The use of accelerator mass spectrometry to obtain early human ADME/PK data[J]. J Expert Opin Drug Metab Toxicol, 2005, 1(1): 23-31.
[3] Higashi T, Shimada K, Toyooka T. Advances in determination of vitamin D related compounds in biological samples using liquid chromatography-mass spectrometry: a review[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2010, 878(20): 1654-1661.
[4] Van den Broek I, Sparidans RW, Schellens JH, et al. Quantitative bioanalysis of peptides by liquid chromatography coupled to (tandem) mass spectrometry[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2008, 872(1-2): 1-22.
[5] Mesaros C, Lee SH, Blair IA. Targeted quantitative analysis of eicosanoid lipids in biological samples using liquid chromatography-tandem mass spectrometry[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2009, 877(26): 2736-2745.
[6] Haleem JI, Josip B. Electrophoresis and liquid chromatography/tandem mass spectrometry in disease biomarker discovery[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2009, 877(13): 1222-1228.
[7] Kostiainen R, Kauppila TJ. Effect of eluent on the ionization process in liquid chromatography-mass spectrometry[J]. J Chromatogr A, 2009, 1216(4): 685-699.
[8] Aubry AF. LC-MS/MS bioanalytical challenge: ultra-high sensitivity assays[J]. J Bioanalysis, 2011, 3(16): 1819-1825.
[9] Deng ZP, Zhong DF, Meng J, et al. Covalent protein binding and tissue distribution of houttuynin in rats after intravenous administration of sodium houttuyfonate[J]. J Acta Pharmacol Sin, 2012, 33(4): 568-576.
[10] Zhou JL, Qi LW, Li P. Herbal medicine analysis by liquid chromatography/time-of-flight mass spectrometry[J]. J Chromatogr A, 2009, 1216(44): 7582-7594.
[11] 范亚新, 陈笑艳, 马智宇, 等. 液相色谱- 串联质谱法以锂加合离子定量分析大鼠血浆中紫杉醇和羟基代谢物[J]. 质谱学报, 2013, 34(3): 137-144.
[12] 芦春梅, 王风红, 胡婷婷, 等. 液相色谱-串联质谱法测定保健品口服液中10种性激素的研究[J]. 化学试剂, 2015, 37(3): 243-246.
[13] C?té C, Bergeron A, Mess JN, et al. Matrix effect elimination during LC-MS/MS bioanalystical method development[J]. J Bioanalysis, 2009, 1(7): 1243-1257.
[14] 刘晓云, 陈笑艳, 钟大放. 液相色谱-串联质谱生物分析方法的基质效应和对策[J]. 质谱学报, 2017, 38(4): 388-389.
[15] Mess JN, Lahaie M, Furtado M, et al. Effect of high pH mobile phase on the sensitivity of multiple drugs by LC positive electrospray ionization MS/MS[J]. J Bioanalysis, 2009, 1(8): 1419-1430.
[16] Wang J, Aubry A, Bolgar MS, et al. Effect of mobile phase pH, aqueous-organic ratio, and buffer concentration on electrospray ionization tandem mass spectrometric fragmentation patterns: implications in liquid chromatography/tandem mass spectrometric bioanalysis[J]. J Rapid Commun Mass Spectrom, 2010, 24(22): 3221-3229.
[17] Torres-Lapasió JR, García-Alvarez-Coque MC. Levels in the interpretive optimisation of selectivity in high-performance liquid chromatography: a magical mystery tour[J]. J Chromatogr A, 2006, 1120(1-2): 308-321.
[18] 李建旺, 王宛, 黃韦, 等. 药代研究中MAS法、蛋白沉淀法和固相萃取法等前处理方法的比较[J]. 质谱学报, 2009, 30(增刊): 128-130.
[19] Michopoulos F, Edge AM, Hui YT, et al. Extraction methods for the removal of phospholipids and other endogenous material from a biological fluid[J]. J Bioanalysis, 2011, 3(24): 2747-2755.
[20] Deng P, Zhan Y, Chen X, et al . Derivatization methods for quantitative bioanalysis by LC-MS/MS[J]. J Bioanalysis, 2012,4(1): 49-69.
[21] Iwasaki Y, Nakano Y, Mochizuki K, et al. A new strategy for ionization enhancement by derivatization for mass spectrometry[J]. J Chromatogr B Analyt Technol Biomed Life Sci, 2011, 879(17-18): 1159-1165.
