三明治结构graphene-2Li-graphene的储氢性能*
2019-03-26周晓锋方浩宇唐春梅
周晓锋 方浩宇 唐春梅
(河海大学理学院, 南京 210098)
本文使用密度泛函理论中的广义梯度近似对扩展三明治结构graphene-2Li-graphene的几何结构、电子性质和储氢性能进行计算研究. 计算得知: 位于单层石墨烯中六元环面心位上方的单个Li原子与基底之间的结合能最大(1.19 eV), 但小于固体Li的实验内聚能(1.63 eV), 然而, 在双层石墨烯之间的单个Li原子与基底的结合能增加到3.41 eV, 远大于固体Li的实验内聚能, 因此位于双层石墨烯之间的多个Li原子不会成簇,有利于进一步储氢. 扩展三明治结构graphene-2Li-graphene中每个Li原子最多可以吸附3个H2分子, 储氢密度高达10.20 wt.%, 超过美国能源部制定的5.5 wt.%的目标. 该体系对1—3 个H2分子的平均吸附能分别为 0.37, 0.17和 0.12 eV, 介于物理吸附和化学吸附 (0.1—0.8 eV)之间, 因此该体系可以实现常温常压下对H2的可逆吸附. 通过对态密度分析可知, 每个Li原子主要通过电场极化作用吸附多个H2分子. 动力学和巨配分函数计算表明graphene-2Li-graphene结构对H2分子具有良好的可逆吸附性能. 该研究可以为开发良好的储氢材料提供一个好的研究思路, 为实验工作提供理论依据.
1 引 言
在当前能源危机越演越烈、环境污染越来越严重的情况下, 高效清洁可再生能源氢能由于具有无污染和可循环使用等优点引起了广泛关注. 氢能在实际使用中面临的一个关键问题就是氢存储. 发展高性能储氢材料是解决氢存储的主要途径. 然而,寻找在室温条件下稳定且有较高储氢密度的材料是一个巨大的挑战. 美国能源部提出2017年实现的储氢材料的储氢密度目标是5.5 wt.%[1,2]. 一般来说, 理想的储氢材料对H2分子的平均吸附能(average adsorption energy, Ead)应介于化学吸附和物理吸附之间(0.1—0.8 eV)[1,2].
目前, 碳纳米结构由于具有较大的比表面积被认为是较有前途的储氢材料. 然而, 纯碳纳米材料由于与H2分子之间存在弱相互作用并不能有效储氢. 因此, 研究者提出了多种途径来改善它们的储氢性能. 其中, 采用金属原子对碳纳米材料进行修饰可以有效增强材料对氢的吸附强度[3-5], 但金属原子在纳米结构表面容易形成团簇, 致使不能有效储氢. 因此, 金属原子与基底之间的结合能(binding energy, Eb)应大于固体金属的实验内聚能[6]. 据悉, 碱金属Li是体积最小、质量最轻的金属, 采用Li原子对纳米材料进行掺杂后可以在更大空间内吸附较多的H2分子进而增加储氢密度.据报道, Li原子与含有Stone-Wals缺陷的石墨烯之间的结合能是2.16 eV[7], 高于固体Li的实验内聚能 (1.63 eV). 另外, Li掺杂的石墨炔[8]、石墨烯[9]和碳纳米管[10]等都表现出良好的储氢性能.实验上也已经证明使用Li原子掺杂的纳米材料可以有效储氢, 例如Li12C60[11]和Li掺杂的多孔材料[12,13]等都是良好的储氢材料.
C6H6-metal-C6H6和C5H5-metal-C5H5等三明治结构由于具有较好的稳定性已经引起了人们的特别关注, 例如: 三明治结构二茂铁(C5H5-Fe-C5H5)[14,15]已被实验合成, 结构中每个C5H5环有5 个电子, Fe 原子的电子结构为 [Ar]3d64s2, 最外层有 8个未配对电子, 因此, 二茂铁结构共有18个电子, 满足18电子规则[16], 形成类似惰性气体的稳定电子组态, 因此能被实验合成. 此外,Lein 等[17]对三明治结构 [E5-Ti-E5]2–(其中 E =CH, N, P, As, Sb)进行了理论研究, 得知满足18电子规则的过渡金属掺杂三明治结构最稳定.但是, 如何让其他种类金属掺杂的三明治结构也稳定下来呢?本文提出C6H6-Li-C6H6结构[18]可以通过吸附H2分子进一步稳定自身的电子结构, 成为良好的储氢体系.
众所周知, 石墨烯是以六元环为基本单位扩展形成的二维平面结构, 目前研究者已经对金属修饰的石墨烯的储氢性能开展了广泛研究, 认为多个金属原子在石墨烯表面上吸附时容易发生聚合现象,使得体系的储氢性能急剧下降[19]. 为了进一步提高金属原子在石墨烯表面上的结合能, 研究者提出了多种方案, 比如在石墨烯中除去一个或者多个C原子形成空位, 使得金属原子在空位处的结合强度大大增强[20]. 如何进一步增强Li原子与石墨烯之间的结合强度是一个需要迫切解决的问题, 对储氢至关重要. 本文提出由C6H6-Li-C6H6扩展形成的graphene-2Li-graphene三明治结构, 并对它的几何结构、电子性质和储氢性能进行计算研究, 第2部分给出计算方法, 结果和讨论在第3部分给出,最后在第4部分给出结论.
2 计算方法
本文中的计算主要采用Dmol3软件包完成[21].计算过程使用密度泛函理论(density functional theory, DFT)[22]中 的 广 义 梯 度 近 似 方 法(generalized gradient approximation, GGA). 选取PBE (Perdew-Burke-Ernzerhof)交换关联梯度修正和极化函数扩展的双数值原子轨道DNP基组. DNP基组就是函数中包含高于自由原子中的最高占据轨道的一级角动量, 与G6-31(d, p)基组的精度一致[23]. 本文对 C, H, Li采用全电子的核处理方式. 众所周知, GGA容易低估体系的结合能, 而局域密度近似 (local density approximation,LDA)又会高估体系的结合能, 因此, 计算中采用了范德瓦耳斯 (van der Waals, vdW)修正 (标准的DFT计算基础上进行势能修正, 即DFT-D方法). 本文主要采用交换关联势中的Grimme扩散修正 (PBE-Grimme[24])方法. PBE-Grimme已经被用于计算多种材料的储氢性能, 比如Pd掺杂的石墨烯[25,26]、C48B12 和 C60Ca32[27]等. 几何结构优化以能量、梯度和位移是否收敛为依据, 自洽场过程以体系的能量和电荷密度是否收敛为依据. 文中对所有的稳定结构都进行了频率计算, 确定无虚频.
为了验证本文计算方法的正确性, 首先对苯环 C6H6进行优化, 得知 C—H键长为 1.09 Å,C—C键长为1.40 Å, 最高占据分子轨道(highest occupied molecular orbital, HOMO)和最低未占分子轨道 (lowest unoccupied molecular orbital,LUMO)之间的能隙 (energy gap, Eg)为 5.20 eV,与 Park等[27]计算得到的 C—H键长 (1.08 Å)、Abad等[28]计 算 得 到 的 C—C键 长 (1.40 Å)、C—C实验键长(1.39 Å[29])和Toyoda等[30]计算得到的能隙值(4.70 eV)非常接近, 以上结果说明本文使用的计算方法可靠.
3 结果与讨论
首先对单个Li原子在C6H6上方的最稳定位置进行研究. 分析可知, 苯环上方有3种不等价位置, 分别为: 1) C 原子顶位上方; 2) C—C 键桥位上方; 3) 六元环面心位上方, 如图 1(a)所示. 计算得知, 三种同分异构体的优化结构中, Li原子都稳定于面心位, 与苯环中心距离为1.78 Å, 如图1(b)所示. Li原子与基底的结合能 binding energy(Eb)定义如下[14,31]:
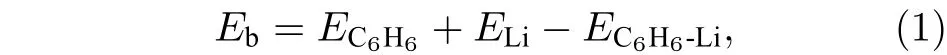
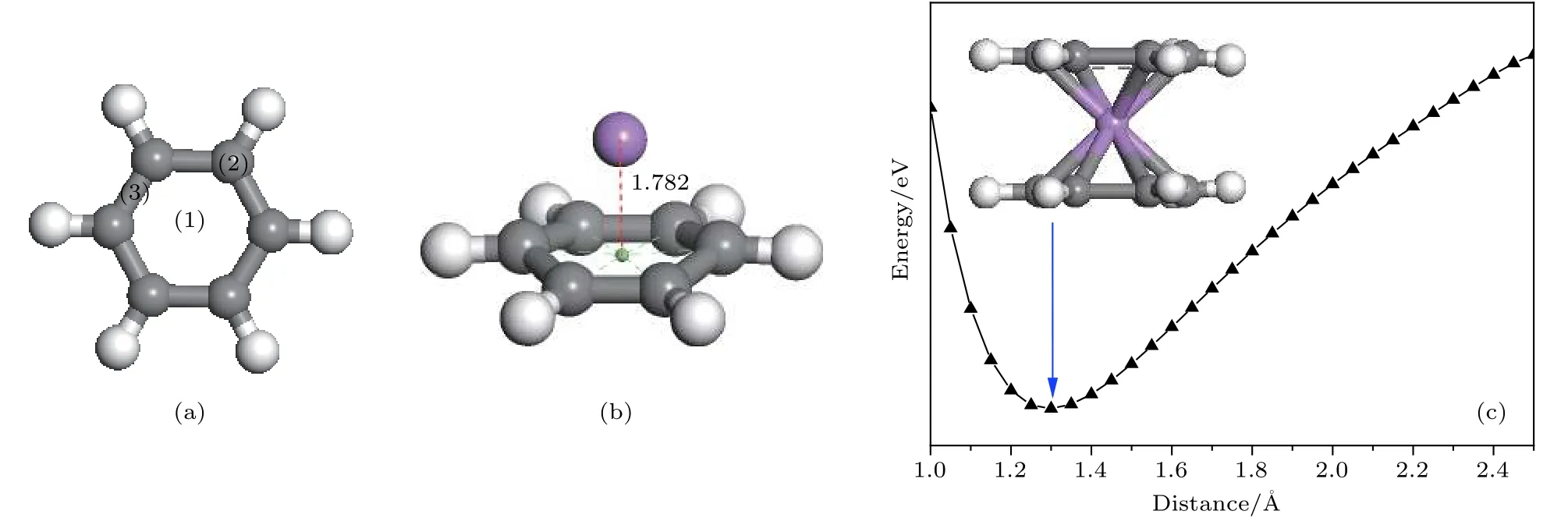
图1 (a)苯环中 3 种不等价位置; (b) Li原子位于苯环面心位上方的优化结构; (c) C6H6-Li-C6H6 势能面扫描曲线和最稳定的C6H6-Li-C6H6三明治结构Fig. 1. (a) Three unequal positions in benzene ring; (b) the optimized structure of the benzene ring with the Li atom located above the face site of the hexagonal ring; (c) potential energy surface scanning curve of C6H6-Li-C6H6 and the most stable sandwich structure of C6H6-Li-C6H6.
Chen等[10]研究发现三明治结构 C6H6-Sc-C6H6比C6H6-Sc结构更稳定, 因此本文接下来进一步将C6H6-Li扩展成三明治结构C6H6-Li-C6H6,并研究其稳定性. 为了得到Li原子在双层石墨烯之间掺杂时的最佳层间距, 首先对 C6H6-Li-C6H6结构的总能量进行扫描, 扫描曲线如图1(c)所示, 以Li原子到苯环平面的距离为变量, 扫描步长为 0.1 Å. 由图可知, Li与下层苯环相距 1.30 Å时, 体系总能量最低. 计算得知, C6H6-Li-C6H6中Li原子与基底的结合能为3.43 eV, 明显大于固体Li的实验内聚能 (1.63 eV)[7], 是 C6H6-Li结构中Li与C6H6之间结合能的3倍多, 因此可以进一步推测, 当上下两个C6H6分别扩展成两层石墨烯平面时, 多个Li原子在双层石墨烯之间进行掺杂时不会发生成簇.
在图 2(a)中, 单层石墨烯的 2 × 3 晶胞中一共有6个六元环, 6个面心位分别定义为 ①—⑥,本文接下来先研究2个Li原子位于平面上的最稳定结构. 根据2个Li原子之间的相对位置, 一共有4种不同组合来放置Li原子, 分别为①②组合、①④组合、①⑤组合和①⑥组合, 它们的优化结构如图 2(b)—图 2(e)所示. 在这四种结构中, Li原子与石墨烯之间的平均结合能(average binding energy, Eavb)定义为石墨烯和Li原子各自的能量之和减去Li掺杂体系的总能量得到的差再除以Li原子数目的值. 计算得知, Li原子与基底的平均结合能分别为 1.38, 1.37, 1.36 和 1.35 eV, 均小于固体Li的实验内聚能(1.63 eV)[7], 表明仍不能有效避免多个Li原子的成簇.
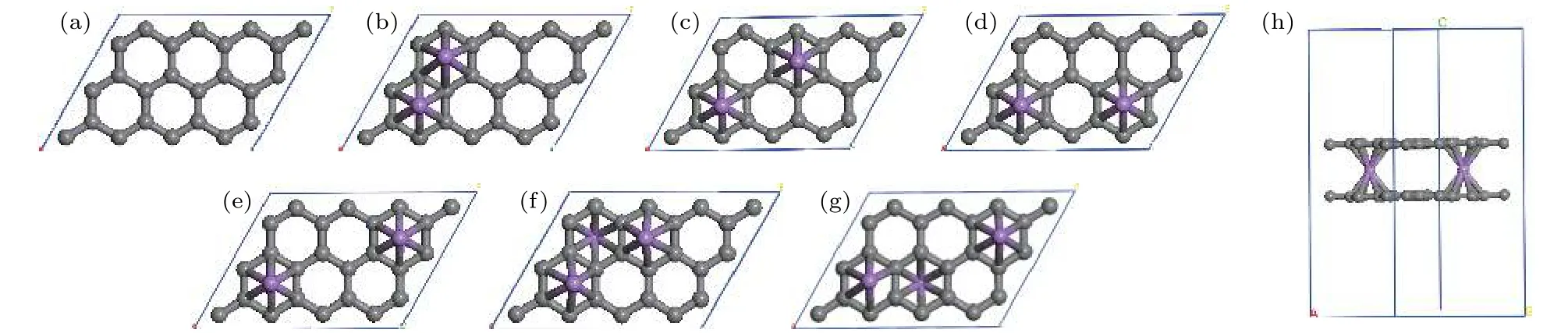
图2 (a) 单层石墨烯的 2 × 3 晶胞中 Li原子的 6 个位置; 2 个 Li原子分别位于 (b) ①④组合; (c) ①⑤组合; (d) ①③组合;(e) ①⑥组合; 3 个 Li原子分别位于 (f) ①④⑤组合; (g) ①②⑥组合; (h) 2 个 Li原子掺杂的最稳定双层石墨烯结构 graphene-2LigrapheneFig. 2. (a) 6 positions of the Li atom in the 2 × 3 unit cell of monolayer graphene; two Li atoms are located at (b) ①④combination; (c) ①⑤ combination, (d) ①③ combination; (e) ①⑥ composition; three Li atoms are located in (f) ①④⑤combination; (g) ①②⑥ combination; (h) the most stable graphene-2Li-graphene double-layer graphene structure doped by two Li atoms.
同时, 当2个Li原子位于石墨烯的同一侧时,石墨烯会向Li原子所在的一侧发生平面弯曲, 这在周期性结构中是一种较不稳定的现象. 为了改善弯曲的石墨烯平面, 在石墨烯另一侧添加第3个Li原子, 分别位于④和②位置, 如图2(f)和图2(g)所示, 图2(f)和图2(g)结构中Li与基底的平均结合能分别为 1.42 和 1.44 eV, 都小于固体 Li的实验内聚能(1.63 eV)[7], 表明多个Li原子吸附在单层石墨烯表面上时会发生成簇.
接下来在双层石墨烯之间放置2个Li原子.根据2个Li原子之间的相对位置, 仍然考虑4种不同组合的吸附结构, 分别为①②组合、①④组合、①⑤组合和①⑥组合. 在优化结构中Li与基底之间的结合能分别为 3.30, 3.35, 3.41 和 3.43 eV,均大于固体Li的实验内聚能(1.63 V)[7], 所以当两个Li原子位于双层石墨烯之间时不会成簇. 其中,两个Li原子为①⑥组合时与基底的平均结合能最大, 因此该结构最稳定, 如图 2(h)所示. 我们注意到, 优化结构中Li原子稍稍向外偏移, 说明它们之间有排斥作用, 因此, 两个Li原子的距离相对较大(①⑥组合)时最稳定, 和平均结合能计算结果一致. 同时计算发现, 图2(h)结构(graphene-2Ligraphene)中 Li与基底的结合能和 C6H6-Li-C6H6中苯环与Li的结合能相同, 说明当苯环扩展成石墨烯平面, 双层石墨之间如①⑥组合放置Li原子时, 上下两层石墨烯对Li原子的结合能基本不会受到影响. 但是, 在图2(h)结构(graphene-2Li-graphene)中两层石墨烯之间的距离增加到3.69 Å (如表 1 所列), 比 C6H6-Li-C6H6结构中两层苯环的距离 (2.60 Å)大出了 0.89 Å, 说明当苯环扩展成石墨烯平面时, 层间放置的Li原子会排斥双层石墨烯在垂直于平面的方向上向外移动.graphene-2Li-graphene结构中Li原子之间的距离为 5.71 Å, 远大于 Li—Li二聚体的键长 (2.61 Å),结合双层石墨烯对Li原子的结合能远大于相邻Li原子之间结合能的事实, 证明多个Li原子位于双层石墨烯之间时不会发生成簇, 有利于进一步储氢.
因为储氢材料必须具有一定的动力学稳定性,我们进一步对图2(h)结构(graphene-2Li-graphene)进行了300 K温度下的动力学模拟. 本文使用Nose-Hoover Chain thermostats进行有限的温度调节, 研究体系的动力学稳定性. 在 300 K, 1 fs步长, 5 ps后的动力学结构与图2(h)结构基本上没有改变, 因此, graphene-2(Li-3H2)-graphene 结构具有较高的热力学和动力学稳定性.
对于已实验合成的二茂铁(C5H5-Fe-C5H5)[14,15]结构, Fe原子的外层电子排布为[Ar]3d64s2, 每个C5H5单元具有5个电子, Fe原子的最外层3d轨道有8个电子, 因此, 该体系一共具有18个电子,形成类似惰性气体的稳定电子态, 因此以二茂铁结构为代表的过渡金属掺杂的三明治结构的稳定性可以使用18电子规则[32]进行解释. 那么, 如C6H6-Li-C6H6等碱金属掺杂的三明治结构如何能稳定下来呢?我们认为在该结构中吸附H2分子是一个很好的途径, 从该思路出发, 碱金属掺杂的三明治结构显然可以成为良好的储氢体系. 接下来就对扩展三明治结构graphene-2Li-graphene的(2 ×3)单元的储氢性能进行深入研究. 图 3(a)—图3(d)分别给出了graphene-2Li-graphene体系的(2 × 3)单元中每个Li原子吸附1—4个H2分子后的优化结构. 表1给出了体系对H2的平均吸附能 Ead、连续吸附能 (consecutive adsorption energy, Ec)、和 Li原子的 Bader电荷 (QLi和 QH)、双层石墨烯的层间距DG-G.
体系对H2分子的Ead定义为[33]
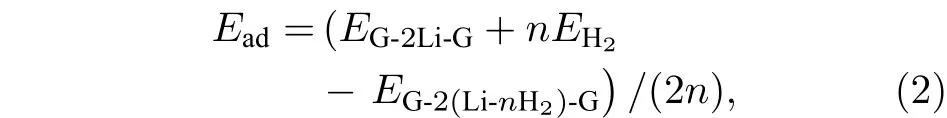
式中 EG-2Li-G,,分别是 graphene-2Li-graphene、单个 H2分子、和 graphene-(LinH2)-graphene 的总能量. 从表 1 可知, graphene-2Li-graphene中每个Li原子对1—3个H2分子的平均吸附能位于0.10—0.80 eV之间, 在能够实现可逆吸附能量值范围内[1,2], 因此, graphene-2Ligraphene中的每个Li原子最多吸附3个H2分子,储氢密度可达10.20 wt.%, 超过美国能源部对储氢材料制定的2017年储氢密度5.5 wt.%的目标[3].
体系对H2分子的连续吸附能(Er)定义为[33]
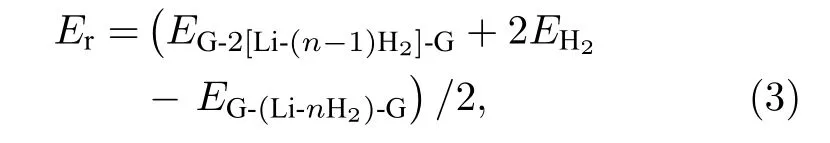
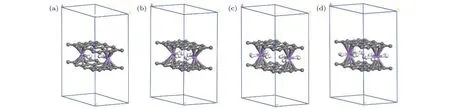
图3 graphene-2Li-graphene 的 2 × 3 晶胞中每个 Li原子分别吸附 1—4 个 H2分子的结构图Fig. 3. The structural of the 2 × 3 unit cell of graphene-2Li-graphene with each Li atom adsorbed by 1−4 H2 molecules.
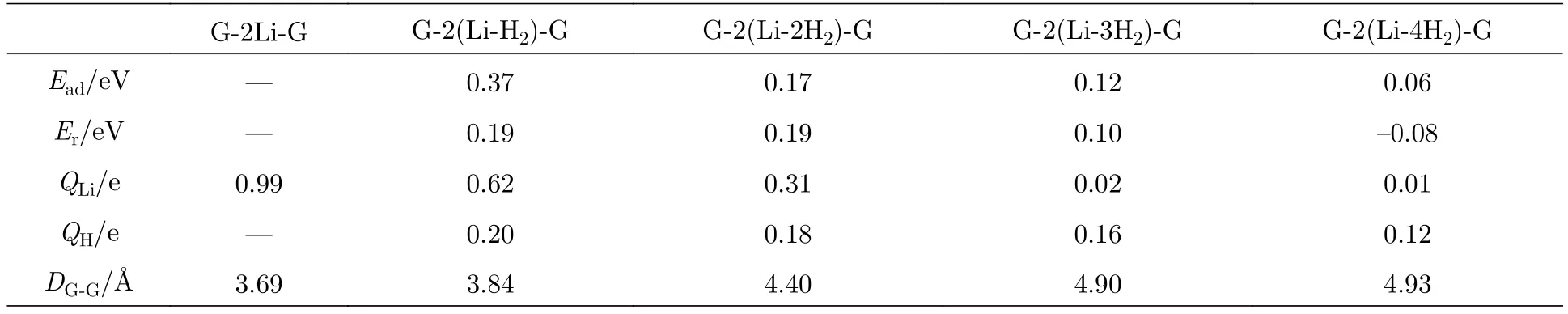
表1 扩展三明治结构 graphene-2(Li-nH2)-graphene[G-2(Li-nH2)-G)](n = 1—4)中的 H2 分子的 Ead, Er, Li和 H 的平均bader电荷(QLi和QH), 双层石墨烯的层间距(DG-G)Table 1. The Ead and Er of H2 molecules average bader charge of Li and H (QLi and QH), interlayer distance of double-layer graphene (DG-G) in the expanded sandwich structure graphene-2(Li-nH2)-graphene[G-2(Li-nH2)-G)](n = 1—4).
由表1给出的Bader电荷可知Li原子带正电荷, 说明Li原子与石墨烯结合时, 会将电荷转移给石墨烯而处于缺电子状态, 所以可以有效吸附H2分子, 而Li原子吸附1—4个H2时每个H原子分别带有平均为 0.20e, 0.18e, 0.16e 和 0.12e 的正电荷, 所以, 电荷应该从H2转移到Li原子最外层的空轨道上, 使得Li原子的最外层轨道逐步被填满从而达到饱和状态稳定下来.
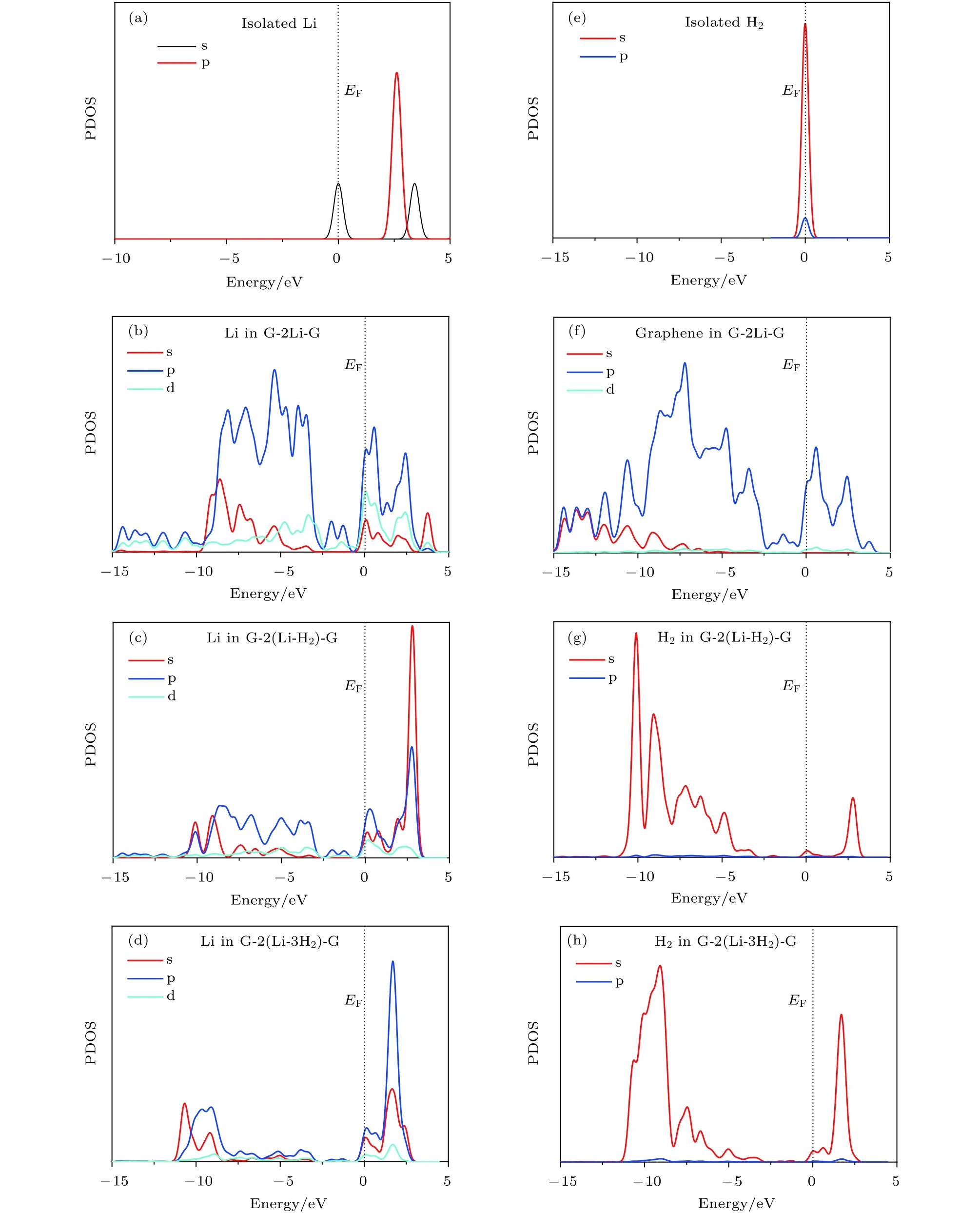
图4 不同结构中 Li原子或 H2 分子的态密度图 (a)单独的 Li原子; (b) graphene-2Li-graphene 中 Li原子; (c) graphene-2(Li-H2)-graphene中 Li原子; (d) graphene-2(Li-3H2)-graphene中 Li原子; (e) 单独的 H2 分子; (f) graphene-2Li-graphene中 graphene;(g) graphene-2(Li-H2)-graphene中 H2分子; (h) graphene-2(Li-3H2)-graphene 中 H2分子Fig. 4. The PDOS of Li atom or H2 molecules: (a) Isolated Li atom; (b) the Li atom in graphene-2Li-graphene; (c) the Li atom in graphene-2(Li-H2)-graphene; (d) the Li atom in graphene-2(Li-3H2)-graphene; (e) isolated H2 molecules; (f) the graphene in graphene-2Li-graphene; (g) the H2 molecules in graphene-2(Li-H2)-graphene; (h) the H2 molecules in graphene-2(Li-3H2)-graphene.
为了进一步理解体系的储氢机理, 本文计算了体系的分态密度 (partial density of states, PDOS),结果如图4所示. 态密度图是通过对体系的离散能级进行洛伦兹展开, 展开系数为 0.15 eV, 由水平轨道给出的权重求和而得到. (HOMO+LUMO)/2被定义为费米能级 (Fermi energy, EF), 在图中取在 0 eV 处, 用黑色虚线表示. 由图 4 可知, 孤立Li原子的 PDOS 主要分布在–2—6 eV 之间, 在费米能级处有两个较宽的峰. Li原子的d轨道态密度分布位于较高能级处, 因此在–15—5 eV的范围内没有被占据. 位于双层石墨烯层间的Li原子的PDOS 的分布扩展到–15—–2 eV 的范围内, 这表明 Li原子的p轨道与石墨烯的C原子的P轨道之间发生轨道杂化, 如图4(b)和图4(f)所示. 每个 Li原子吸附 1个 H2分子后, G-2(Li-H2)-G中H2的PDOS的一个峰分布在–13—5 eV之间,对应于H2的成键态, 进一步说明H—H键未断裂, 如图 4(g)所示. H2分子的成键轨道与 Li原子的 s和 p 轨道的 PDOS 在–13— –2 eV 之间有明显重叠; 在 0—5 eV 范围内, H2分子的 PDOS有几个低峰, 对应于 H2分子的反键轨道, 它与Li原子的s和p轨道之间也存在杂化, 如图4(c)和图 4(g)所示. 结合 Bader电荷可知, Li原子最外层的 1个电子转移给了 C原子, 因此带有0.99e的电荷在其周围激发电场, 通过电场极化作用吸附 H2分子. H2分子的 H—H 键因此变长, 吸附作用得到加强. G-2Li-G中Li原子的p轨道主要分布在–15—5 eV 之间, 较大的峰位于–10— –5 eV之间, 而吸附H2分子之后移到了费米能级之上的0—5 eV 之间, Li原子所带的电荷越来越少, 说明Li原子靠近费米能级处的空轨道逐渐被来自H原子的电子占据, 形成较大的电子占据态, 如图4(d)和图4(h)所示. 综上所述, 三明治结构graphene-2Li-graphene中每个Li原子主要通过电场极化作用吸附1—3个H2分子.
为了研究储氢体系是否具有良好的氢释放性能, 接下来对 graphene-2(Li-3H2)-graphene结构在温度77 和300 K下进行动力学模拟. 本文使用Nose-Hoover Chain thermostats进行有限的温度调节, 研究储氢结构的氢释放性能. 图5给出了graphene-2(Li-3H2)-graphene 结构在 77 和 300 K下 5 ps之后的动力学结构图. 由图 5 可知, 结构在 77 K 和 1 fs步长下 5 ps后释放 2 个 H2分子. 而在300 K和1 fs步长下5 ps后释放 5个H2分子.因此, 在较高的温度下, 更多的H2会被释放出来.上述结果表明graphene-2(Li-3H2)-graphene结构对H2具有良好的可逆吸附性能.
进一步根据巨配分函数[34]求得在一定温度下体系能够吸附的平均H2分子数, 在温度T时,graphene-2Li-graphene能够吸附的平均氢分子数由下面的公式计算得到:
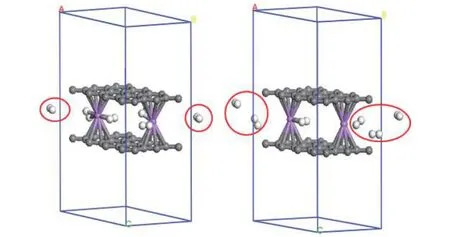
图 5 graphene-2(Li-3H2)-graphene在 77 和 300 K 下5 ps之后的动力学结构图Fig. 5. The structures of graphene-2(Li-3H2)-graphene at 77 and 300 K after 5 ps dynamic times.
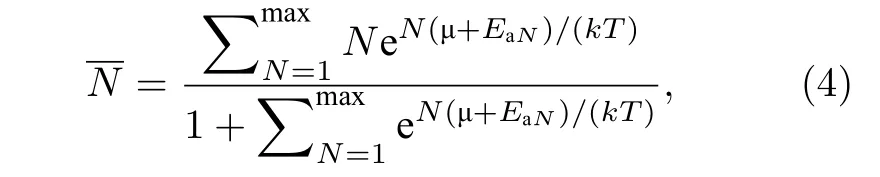
式中k为玻尔兹曼常数, T是开尔文温度, EaN为体系吸附N个H2时的平均吸附能,为一定压强和温度下 H2的化学势. 由 (4)式计算可知, 在1 atm (1 atm = 1.01 × 105Pa), 400 K 的条件下, 每个Li原子能够吸附的平均H2分子数为0.08,接近于 0, 因此, H2几乎完全释放. 因此, Li原子掺杂的三明治结构graphene-2Li-graphene可以实现在常温常压下对H2分子的可逆吸附.
4 结 论
本文主要得到以下几点结论:
1)单层石墨烯中六元环面心位上方是单个Li原子的最稳定位置, 但是Li原子与石墨烯的结合能(1.19 eV)小于固体Li的实验内聚能(1.63 eV),因此当多个Li原子吸附时会发生成簇现象;
2) Li原子在双层石墨烯之间掺杂形成的扩展三明治结构graphene-2Li-graphene中Li原子与双层石墨烯的结合能高达3.43 eV, 远大于固体Li的实验内聚能(1.63 eV), 因此当多个Li原子在双层石墨烯之间掺杂时不会成簇;
3)周期三明治结构graphene-2Li-graphene中每个Li原子最多可以吸附3个H2分子, 储氢密度高达 10.20 wt.%, 超过美国能源部制定的 5.5 wt.%的目标;
4)通过对态密度分析可知, Li原子掺杂的三明治graphene-2Li-graphene主要通过电场极化的作用吸附H2分子;
5)动力学和巨配分函数计算表明, Li原子内掺杂的双层石墨烯结构对H2分子具有良好的可逆吸附性能.
