DIAPH1基因相关性遗传性耳聋研究进展*
2019-03-19吴侃王秋菊
吴侃 王秋菊
常染色体显性遗传性耳聋(autosomal dominant sensorineural hearing loss,ADNSHL)是一种遗传异质性明显的疾病,目前已发现38个基因与之相关(http://hereditaryhearingloss.org)。1992年Leon等[1]在一个哥斯达黎加耳聋家系,利用限制性片段长度多态性(restriction fragment length polymorphisms, RFLP)标记的方法定位了第一个常染色体显性遗传性耳聋基因座位,即DFNA1。1997年Lynch等[2]确定DFNA1型耳聋致病基因为DIAPH1。20年来,已有数个DIAPH1基因突变引起的遗传性耳聋家系被报道,大多数患者表现为迟发性听力损失伴或不伴血液学异常。此外,还发现DIAPH1基因突变与常染色体隐性遗传小头综合征相关。遗传性耳聋具有明显的表型多样性和遗传异质性。以往由于上一代测序技术的局限,耳聋基因及突变的发现无异于大海捞针。近些年来,二代测序技术的发展使基因突变的发现速度大大加快,因此,有必要对一些常见或具有特征性的基因及突变进行梳理和总结。DIAPH1被认为是少数几种与低频听力损失相关的耳聋基因之一[2~5]。本文将从基因的结构、功能及相关临床表型对DIAPH1基因研究进行梳理,以加深对DIAPH1基因型-表型的关联性认识和临床遗传咨询指导。
1 DIAPH1基因的结构
DIAPH1基因(OMIM:* 602121)全称diaphanous related formin 1,其他名称有:DIA1、DRF1、DFNA1、LFHL1、SCBMS、hDIA1。定位于5q31.3,基因组位置chr5:141,515,021-141,619,055,全长104 035 bp,cDNA含28个外显子。由于剪接位点的不同,人类DIAPH1基因有3种不同的转录变异体(transcript variants)(表1),包括isoform 1、isoform 2、isoform 3。其中,isoform 1编码的蛋白质最长,常作为DIAPH1基因编码DNA的标准序列,其mRNA全长5 804 bp,蛋白质编码区域在mRNA的142 bp~3 960 bp区域,编码含有1 272个氨基酸的蛋白质。
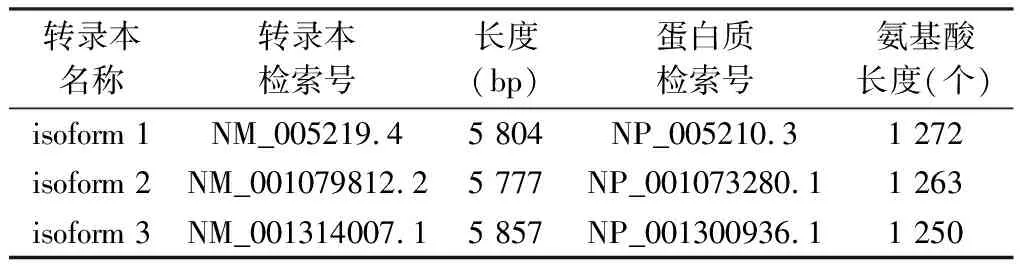
表1 DIAPH1基因的不同转录本检索号及长度和其编码的蛋白质及氨基酸*
注:*来源于https://www.ncbi.nlm.nih.gov/
DIAPH1基因编码的蛋白质diaphanous homolog 1属于diaphanous相关成蛋白家族(diaphanous-related formins),目前已发现了该家族的3个亚型(DIAPH1、DIAPH2、DIAPH3),其中DIAPH2与X-连锁的卵巢功能早衰相关[6],DIAPH3与听神经病相关[7],也有文献[8~10]报道其通过重构细胞骨架来调节细胞的运动,从而与肿瘤细胞的侵袭转移相关。而DIAPH1是一种多结构域蛋白(图1),包含GBD结构域(diaphanous GTPase-binding domain),长度184个氨基酸残基(84~268);FH3/DID结构域(diaphanous FH3 domain/ diaphanous inhibitory domain),长度为192个氨基酸残基(274~466);FH1结构域(formin homology region 1),长度为148个氨基酸残基(616~764);FH2结构域(formin homology 2 domain),长度为437个氨基酸残基(770~1 207);DAD结构域(diaphanous auto-regulatory domain),长度为28个氨基酸残基(1 194~1 222)。目前认为CC(coiled-coil)结构域(470~550)参与了FH2二聚体的形成[11];FH3是否作为一个独立的有作用的结构域仍然有待研究[12];DID结构域(129~369)是FH3结构域上一段靠近N-末端的氨基酸肽链,不容易被蛋白酶所降解[13],DAD结构域(1 194~1 222)并不是严格意义上的结构域,而是一段FH2结构域C末端侧的20~30个氨基酸序列[14]。
2 DIAPH1基因的功能研究
DIAPH1基因在肌动蛋白微丝和微管细胞骨架组装过程中起到重要的调节作用,其蛋白是Rho小GTP结合蛋白的效应蛋白[12~14]。静止状态下C-末端的DAD结构域与N末端的FH3/DID结构域相互作用,DIAPH1蛋白处于自抑制状态。而Rho小GTP结合蛋白通过占据DIAPH1蛋白N末端的GBD结构域的结合部位[13~15],在某些共同作用因子(co-facters)的协助下分两步解除了自抑制作用[16, 17]。FH1结构域以多聚脯氨酸序列为特征,是profilin(肌动蛋白单体结合蛋白)的结合位[18~21]。解除自抑制作用后的FH2结构域在结合了profilin蛋白的FH1结构域帮助下,将肌动蛋白的单体连接到肌动蛋白微丝barbed末端[22],促进了聚集成核、延伸、加帽及微丝延长等过程,最终对细胞的粘附、迁移,对损伤和感染做出应答的过程中发挥重要的调控作用[23~25]。
3 DIAPH1基因突变及其相关表型
1997年以来,共有来自哥斯达黎加、韩国、德国、英国、法国、日本等6个国家的学者报道了该基因的6个杂合突变与常染色体显性遗传性迟发性聋相关。本文根据王秋菊等[26]翻译整理的2015年ACMG指南和遗传变异致病性评价的相关文献[27]对这些变异致病性进行重新评价(表2),并总结DIAPH1蛋白结构上的突变定位(图1),对临床表型进行总结梳理(表3、4)。
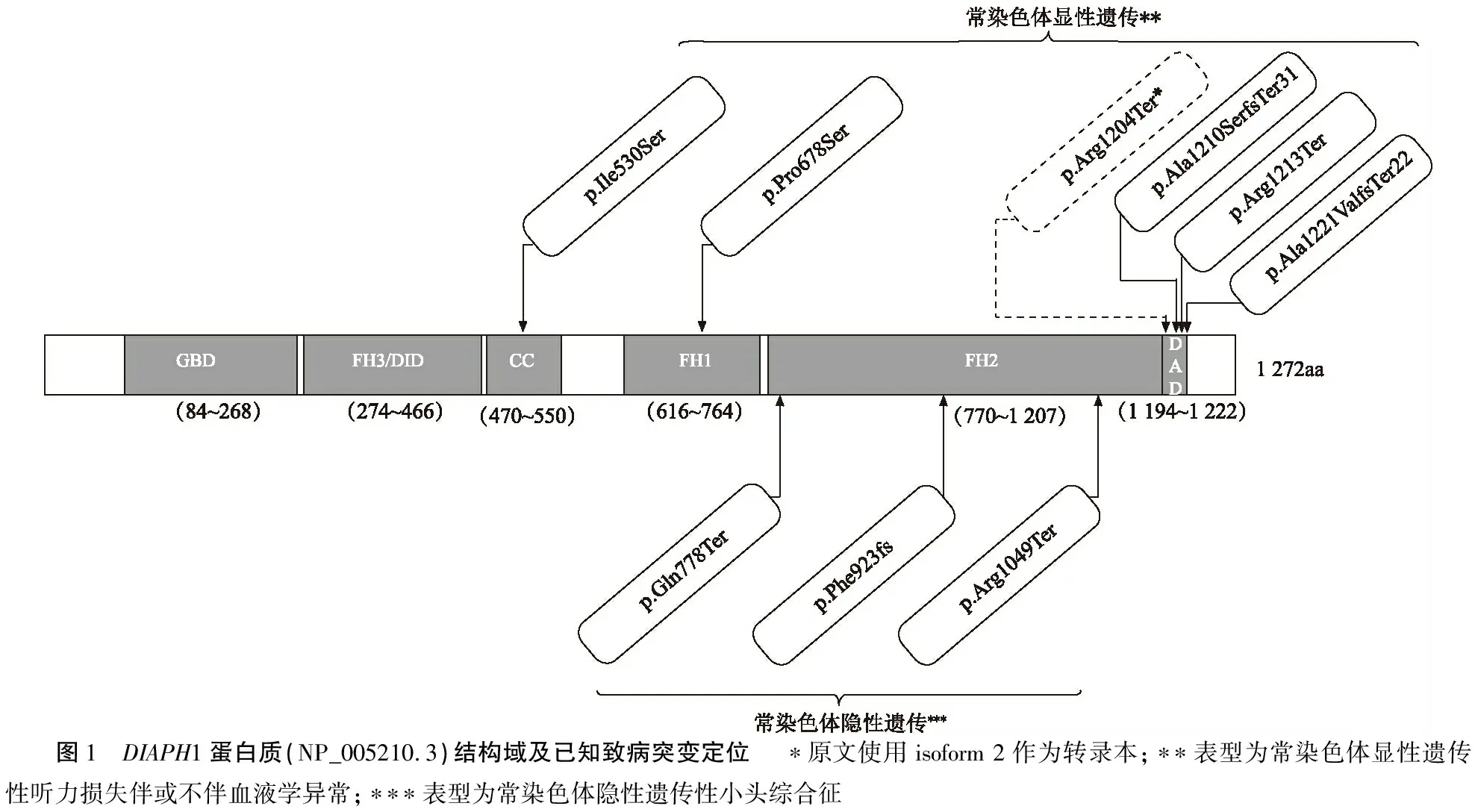
图1 DIAPH1蛋白质(NP_005210.3)结构域及已知致病突变定位 ∗原文使用isoform 2作为转录本;∗∗表型为常染色体显性遗传性听力损失伴或不伴血液学异常;∗∗∗表型为常染色体隐性遗传性小头综合征
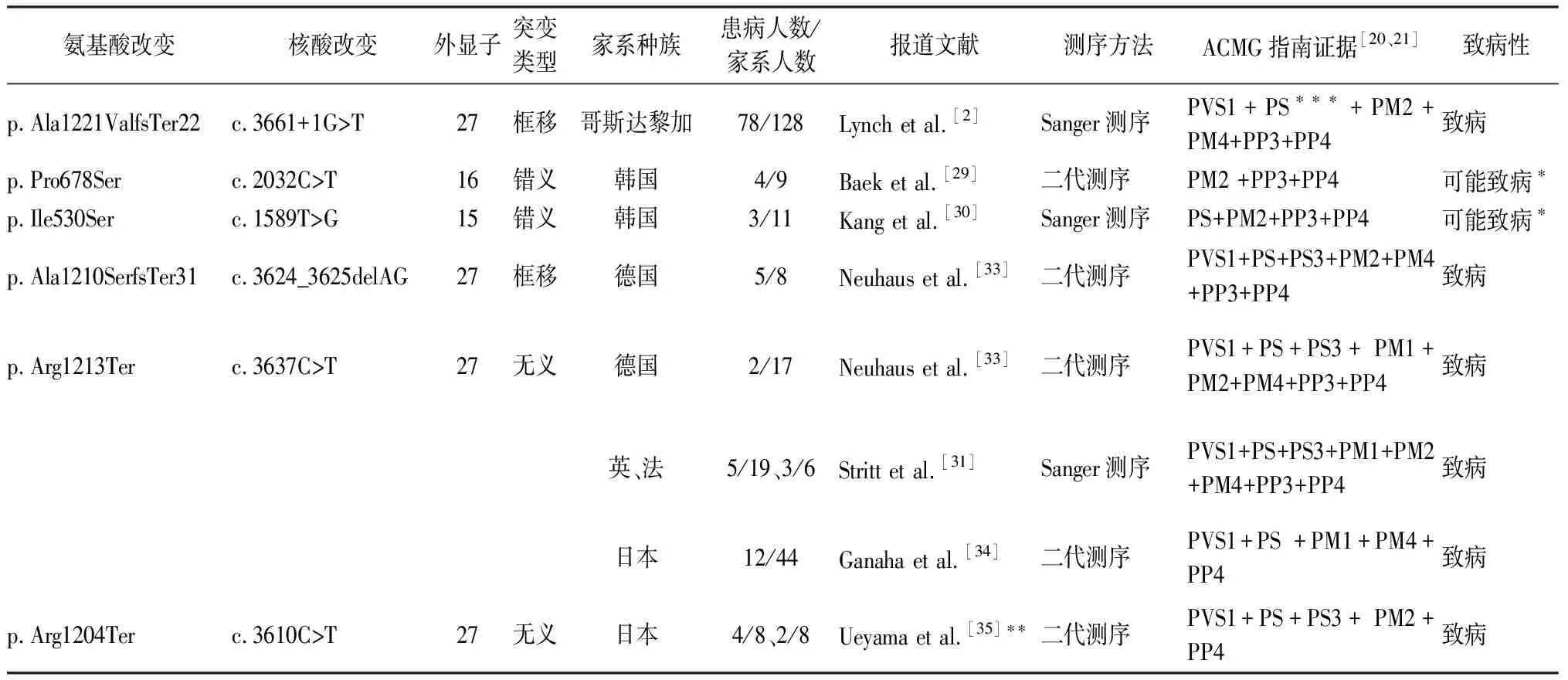
表2 已报道的常染色体显性遗传DIAPH1突变特征及ACMG指南致病性分析
注:*原文中DIAPH1突变致病性证据按ACMG指南的标准并不充分,划为可能致病;**原文转录本为isoform 2(NM_001079812.2),余文献转录本为isoform 1(NM_005219.4);***在ACMG指南中,共分离证据的强度是PP到PS逐渐增加的,但并没有给予相应的编号,故此处以PS表示强的共分离证据。
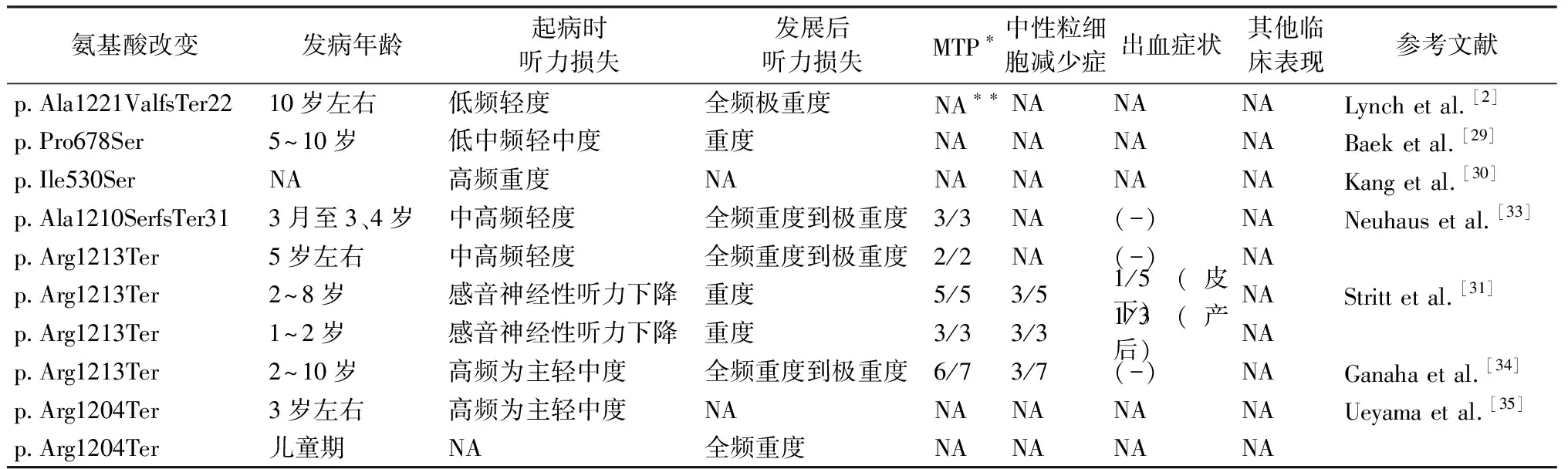
表3 已报道的常染色体显性遗传DIAPH1突变的氨基酸改变及其表型特征
注:*MTP(macrothrombocytopenia,巨血小板减少症);**NA文中未提及
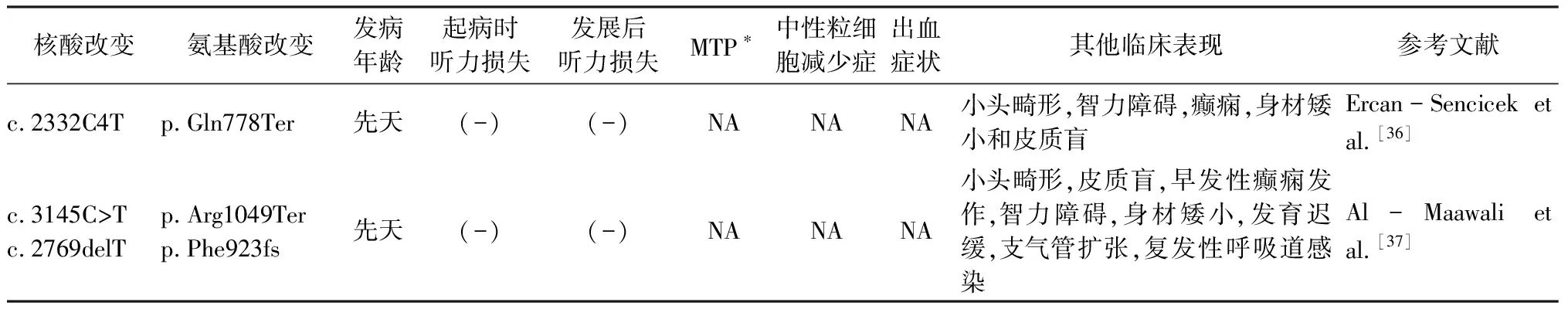
表4 已报道的常染色体隐性遗传DIAPH1突变及其表型特征
注:*MTP(macrothrombocytopenia,巨血小板减少症);**NA文中未提及
3.1常染色体显性遗传非综合征型耳聋 1997年Lynch等[2]报道了1个8代相传常染色体显性遗传性耳聋的哥斯达黎加大家系(M家系),利用家系连锁分析、Sanger测序和定位克隆技术,将DFNA1型耳聋基因定位于5q31.3的DIAPH1基因;其发现该家系中耳聋患者DIAPH1基因倒数第2个外显子存在一个G到T的替换,使剪切位点后移,并插入4 bp(TTAA)的碱基,造成下游序列框移突变和翻译提前终止;突变基因翻译出的蛋白质含有21个异常氨基酸,尾部缺失32个氨基酸;家系中的患者多为10岁左右发病,起病时为低频听力损失,逐渐发展,至30岁左右发展为双侧全频极重度听力损失。随后Lalwani等[28]对该家系患者表型进行了进一步的描述,并推测内淋巴积水有可能是该家系患者出现低频听力损失的机制之一。受制于测序方法的局限性,随后15年并没有再发现DIAPH1相关性DFNA1型耳聋,直到2012年Baek等[29]利用新一代测序技术,在韩国8个小家系中发现了5种已知耳聋基因的罕见突变位点,其中之一为DIAPH1基因的错义突变c.2032 C>T(p.Pro678Ser);该突变位于16号外显子,造成位于FH1结构域的多聚-L-脯氨酸延伸结构上的疏水的脯氨酸变为亲水的丝氨酸,作者猜测其可能对肌动蛋白聚合产生影响,最终扰乱耳蜗毛细胞的极性;家系中患者表型符合DIAPH1相关性耳聋低频听力损失的特点,多数5~10岁发病,无其他伴随症状。但参照ACMG遗传变异分类标准与指南[26],该家系中两代4名成员共分离,计算得知N=1/2×1/2=1/4,BF=3.99~1/N,达不到有效的共分离证据的标准[27],因此,在文中只能找到1个PM2、1个PP3、1个PP4的证据,该突变只能被列为可能致病突变。同样是错义突变,2017年Kang等[30]报道的DIAPH1新突变c.1589T > G(p.Ile530Ser),因家系较大,可得出强的共分离证据PS(强的共分离证据在ACMG指南中没有相对应的PS编号),文中共找到1个PS、1个PM2、1个PP3和1个PP4的证据,也仅能得出可能致病突变的结论。该错义突变位于15号外显子,推测非极性的异亮氨酸被极性的丝氨酸所替代,氨基酸极性的变化使CC(coiled-coil)结构域螺旋结构的稳定性降低,是DIAPH1突变致病的新假说,但仍需要进一步的功能研究提供更强烈的致病性证据。
3.2常染色体显性遗传性耳聋伴血液学异常 2016年Stritt等[31]首次发现DIAPH1致病突变c.3637C>T(p.Arg1213Ter)不仅引起感音神经性听力损失,还与巨血小板减少症(macrothrombocytopenia,MTP)相关;MTP表现为血小板体积增大和数目减少,作者将这种综合征样表型命名为DIAPH1-RD(DIAPH1相关性疾病);在电镜下观察可见患者血小板形态出现异常:细长而含颗粒少,球形而颗粒多,细胞质内出现空泡和膜复合物;部分患者可伴有中性粒细胞减少症(neutropenia)和出血倾向(abnormal bleeding symptom);作者推测该突变导致1 213位精氨酸被终止密码子替代,造成翻译提前终止,截短的DIAPH1变异蛋白缺少DAD base RRKR模体(1 213~1 216),导致DAD-DID蛋白复合体无法形成,使DIAPH1丧失自我抑制调节能力;而前血小板的形成过程与上述DIAPH1对肌动蛋白和微管细胞骨架的动力学负调节作用相关[32];但文中并没有详细描述患者听力损失的表型特征。Neuhaus等[33]研究发现位于DIAPH1基因27号外显子的框移突变 p.Ala1210SerfsTer31(c.3624_3625delAG)和无义突变c.3637C>T (p.Arg1213Ter)同样引起了类似的DIAPH1-RD,并且详细分析了患者的听力损失特征,发现与以往DIAPH1突变引起低频听力损失不同,该突变患者表现为先天性中高频听力损失并且快速进展,部分患者伴中性粒细胞减少症和出血倾向。有研究[33]使用免疫荧光技术研究了DIAPH1在小鼠耳蜗细胞及神经结构的分布,发现在耳蜗中的内柱细胞(IPC)、外毛细胞(OHC)基底部(可能为Deiter细胞)、螺旋神经节神经元、蜗神经中心区域以及雪旺细胞和少突胶质细胞之间的交界区域都有DIAPH1的分布;推测上述突变可能影响肌动蛋白和微管的重构,导致内耳细胞和巨核细胞中细胞骨架和刚性结构发生改变,是耳聋伴随巨核细胞血小板减少症的机制之一,因此,建议将血常规检查作为常染色体显性遗传性耳聋患者的的常规检查项目,以帮助锁定具有综合征样表型特征的DIAPH1基因突变。同样的无义突变和类似综合征样表型也见于Ganaha等[34]的报道,该研究提出位于DAD结构域的突变位点c.3637C>T(p.Arg1213Ter)是该基因的热点突变位点;不同的是该文中家系患者的血小板数量比Strilt等[31]的报道更少,且随着年龄增长而减少,因此推测血小板减少也是渐进性的;文中指出MYH9相关性综合征如:May-Hegglin anomaly(OMIM 155100)、Fechtner syndrome(OMIM 153640)、Epstain syndrome(OMIM 153650)、Sebastian syndrome(OMIM 606249)也表现为听力损失及MTP,因此,建议有DIAPH1-RD的患者需要同时检测DIAPH1和MYH9基因。但常染色体显性遗传性耳聋伴血小板减少症分子机制目前尚未明确。
另外,Ueyama等[35]使用不同的转录本isoform 2(NM_001079812.2)在2个日本家系中检出DIAPH1基因c.3610C>T突变(p.Arg1204 Ter),在蛋白质水平突变同样位于DAD结构域,患者表现为儿童期发病,听力损伤为轻中度,以高频为主,迅速发展为双侧全频重度感音性聋;该研究利用DIA1(R1204 Ter)-TG mice(转基因小鼠)构建的动物模型对该突变造成的临床表型、耳蜗细胞形态学改变做了较深入的研究,印证了该无义突变引起翻译过程提前终止,DADbase RRKR(1204-1207)模体的缺失是该突变的作用机制;但该研究并没有做血液学的相关检测。
3.3常染色体隐性遗传性小头综合征 除了常染色体显性遗传导致感音神经性听力损失伴或不伴血液学异常,曾有中东地区的学者[36, 37]报道过3个近亲结婚的家庭成员出现DIAPH1纯合突变相关的小头综合征,可出现小头畸形、智力障碍、癫痫、身材矮小、皮质盲、呼吸道疾病等综合征样表型,但患者及其杂合突变携带者的父母均未出现听力损失。突变造成了DIAPHI蛋白FH2、DAD等重要结构域的缺失,DIAPH1的表达与纺锤体中心体有关,从而影响了神经元细胞的有丝分裂和迁移,最终限制了大脑的发育[36]。Ercan-Sencicek等[36]专门研究了DIAPH1在内耳和神经系统的分布,发现外周神经和听觉中枢、听神经的髓鞘组成部分雪旺细胞和少突胶质细胞中存在大量的DIAPH1分布,推测神经系统髓鞘的缺陷可能是DIAPH1纯合突变引起常染色体隐性遗传性小头综合征的机制之一。至于DIAPH1纯合突变的患者不出现听力损失而存在小头综合征的机制仍然有待研究(突变位点定位见图1,临床表现见表4)。
4 总结和展望
综上所述,DIAPH1基因突变可引起常染色体显性遗传的迟发性感音神经性耳聋伴或不伴血液学异常及常染色体隐性遗传性小头综合征。作为DFNA1型耳聋的致病基因,以往认为DIAPH1基因突变主要与低频遗传性耳聋相关,儿童期发病,30岁左右发展至重度到极重度全频听力损失;近年来一些文献报道该突变也可引起中高频听力损失,体现了常染色体显性遗传性耳聋的表型多样性。但多数文献中家系患者例数较少,受环境等因素影响较大,无法确定听力损失特征的可靠性。既往报道伴血液学异常的DIAPH1相关性耳聋患者多数没有出血等临床症状,血小板数目及大小与DIAPH1基因突变是否有关尚需进一步研究,且MYH9基因相关性综合征型耳聋也可有类似的血液学异常,因此,检测血液指标对于DIAPH1相关性耳聋的诊断及治疗尚没有明确的临床意义。
迄今为止已发现的该基因突变除了2种错义突变位于16号外显子,其余突变均位于第27号外显子,造成框移突变或无义突变,相应突变导致DIAPH1蛋白自抑制相关的DAD结构域(1 194~1 222)重要部分,如:DAD base RRKR motif (1 213~1 216)缺失或使剪切位点后移,造成翻译提前终止和异常蛋白质生成。特别是无义突变c.3637C>T(p.Arg1213Ter)在多个家系中被发现,可能是DFNA1型耳聋的热点突变。根据ACMG指南,c.1589T>G(p.Ile530Ser)和c.2032 C>T(p.Pro678Ser)两种错义突变的致病性尚需进一步功能研究来明确。
近年来,遗传学、分子生物学、细胞生物学、二代测序技术、生物信息学的发展极大地推动了遗传性耳聋致病突变的发现和功能研究。目前只发现一个DIAPH1突变大家系的表型呈现为特征性的低频听力损失,该基因与特征性表型的相关性尚待大家系的发现和深入的基因型-表型分析。该基因与血液学异常和小头综合征的关系仍缺乏深入的基础研究。围绕DIAPH1基因与相关突变的深入研究将帮助人类加深对遗传性耳聋的认识,有利于揭示相关疾病的发病机理、临床表型、发展及预后,为遗传性耳聋的临床诊断,特别是低频听力损失为主的常染色体显性遗传性耳聋的诊断提供参考,为遗传咨询工作和耳聋基因的诊断提供理论依据。
