2型糖尿病对大鼠长骨结构与骨髓间充质干细胞增殖分化的影响*
2019-03-14王译凡贺琦多
胡 巍 刘 娜 王译凡 李 影 贺琦多 郭 斌
糖尿病是以高血糖为特征的慢性代谢性疾病,由胰岛素分泌不足或胰岛素抵抗引起,分为1型、2型、妊娠型及其他如基因缺陷等少数特殊型糖尿病,而其中2型糖尿病最为常见,占比超过90%。2型糖尿病以胰岛素分泌相对不足为特征,可表现为胰岛素抵抗综合征[1]。长期进展的糖尿病会导致多组织器官如眼部、肾脏、骨组织、心血管系统等的慢性损害和功能障碍,其并发症严重影响身体健康[2,3]。
骨形成与骨吸收活动的动态平衡维持了骨组织的正常生长及代谢活动,失衡引起骨组织硬化、骨质疏松及退化吸收等骨疾病。临床及体外细胞学实验表明,1型糖尿病和2型糖尿病均可增加骨折风险及干扰骨愈合[4],且1型糖尿病主要干扰骨形成代谢过程而非骨吸收代谢过程,对成骨进程的相关细胞可表现出抑制作用[5]。然而目前关于2型糖尿病对体内长骨骨组织结构和骨髓间充质干细胞(bone marrow stem cells,BMSCs)的影响研究鲜少展开。本研究拟建立2型糖尿病大鼠模型,分析2型糖尿病对长骨形态参数以及对骨髓间充质干细胞生物学功能的影响,为防治2型糖尿病引起的骨损伤疾病提供依据。
1.材料和方法
1.1 材料
1.1.1 实验动物与分组 健康清洁SPF级雄性GK大鼠和雄性Wistar大鼠各8只,6周龄,体质量GK大鼠138.5±17.1g,Wistar大鼠161.3±15.9g,购自常州卡文斯实验动物有限公司,动物合格证号:201723853。
1.1.2 主要试剂与仪器 α-MEM培养液、胎牛血清(Gibco,美国),青霉素/链霉素混合液、胰蛋白酶-EDTA消化液(HyClone公司,美国),超敏感大鼠胰岛素酶联免疫试剂盒(Mercodia,瑞典),BCIP/NBT底物显色试剂盒(上海碧云天生物技术有限公司),Annexin V-FITC/PI凋亡检测试剂盒、Cell Counting Kit-8试剂盒、DNA含量检测试剂盒(细胞周期),Trizol reagent RNA提取试剂盒(Invitrogen,美国)、PrimeScriptTM RT Reagent Kit(TaKaRa, 日 本 ), TransStart Top Green qPCR SuperMix(北京全式金生物科技有限公司),β-甘油磷酸钠、L-维生素C、地塞米松(Sigma,美国),GEeXploreLocusMicroCT扫描机(美国通用公司),罗氏ACCU-CHEKR Advantage血糖仪(Roche,瑞士),BD FACS流式细胞仪(BD,美国),CFX96实时定量PCR仪(Bio-Rad,美国)。
1.2 方法
1.2.1 构建2型糖尿病大鼠模型 采用GK大鼠纯食物诱导方法构建2型糖尿病模型。自6周龄至14周龄,8只GK组大鼠作为实验组,辅以高脂饲料喂养;8只Wistar大鼠作为对照组,辅以普通饲料喂养。每周于固定时间禁食、不禁水12h后进行生理指标测定,包括体重、空腹血糖,使用罗氏ACCU-CHEKR Advantage血糖仪测定大鼠尾静脉血血糖值。大鼠至14周龄夜间禁食、不禁水12h后,检测各组空腹胰岛素,再进行口服葡萄糖耐量试验(oral glucose tolerance test,OGTT)。胰岛素测定时采血自大鼠内眦静脉,应用超敏感大鼠胰岛素酶联免疫试剂盒检测空腹胰岛素值。口服葡萄糖耐量试验给予50%葡萄糖溶液4.2g/kg灌胃,分别在空腹时、糖负荷30min、60min和120min后于尾静脉采血并测定血糖值。2型糖尿病大鼠模型构建成功的标准:空腹血糖值≥11.1mmol/L或OGTT实验120min后血糖大于16.7mmol/L。
1.2.2 显微CT扫描胫骨结构形态计量学指标选取建模成功的两组GK组大鼠和Wistar组大鼠各8只,注射戊巴比妥钠处死大鼠,解剖双侧股骨和胫骨。咬骨钳去除股骨两端骨质,暴露骨髓腔,利用干细胞培养液冲洗出骨髓,并置于37℃、5%CO2及饱和湿度的环境进行细胞培养;胫骨放置于多聚甲醛固定液固定24h后,采用美国通用公司GE eXplore Locus MicroCT扫描机(voltage 80V,current 450μA,bin mode 1×127μm)对胫骨近端骨骺端进行连续断层扫描。
1.2.3 细胞纯化 待原代细胞汇合至80%时,加入0.25%胰蛋白酶-EDTA消化成细胞悬液,再加入等量细胞培养液终止反应,1000r/min离心5min,弃上清后加入细胞培养液重悬细胞,应用有限稀释法培养BMSCs。调整细胞密度至1.5×104/mL,向96孔板每孔中加入0.1mL细胞悬液,37℃、5%CO2细胞培养箱内培养48h后标记单个细胞孔,补加0.1mL细胞培养基,3日换液一次,待克隆细胞长至96孔板底汇合至60%时,0.25%胰蛋白酶-EDTA消化、扩大培养,用于后续实验。
1.2.4 CCK-8法检测细胞增殖能力 取第三代生长良好的各组骨髓间充质干细胞,用0.25%胰蛋白酶-EDTA消化成细胞悬液。稀释细胞悬液,细胞计数后以5×104/ml浓度的悬液按照100μL/孔接种至96孔板,将培养板放入培养箱预培养,分别在1d、2d、3d、4d、5d后向每孔加入10μL CCK-8溶液,用酶标仪测定各孔在450nm处的吸光度。
1.2.5 细胞凋亡检测试验与细胞周期检测试验收集各组第三代骨髓间充质干细胞1×106/次,用ANNEXIN V-FITC/PI凋亡检测试剂盒中的Binding Buffer悬浮并重悬细胞使细胞密度达到1×106/ml,每管加入100μL细胞,再向管中加入5μL Annexin V-FITC,再加入 5μL PI溶液孵育5min,最后每管加入PBS溶液至500μL,流式细胞仪上机检测;调整第三代骨髓间充质干细胞细胞浓度为1×106/ml,取1ml单细胞悬液,离心单细胞悬液,去除上清液,在细胞中加入70%预冷乙醇500μL,固定2小时4℃保存至过夜,在细胞沉淀中加100μL RNaseA溶液,37℃水浴30min,加入400μL PI染色液混匀孵育30min,流式细胞仪进行上机检测,记录激发波长488nm处红色荧光。
1.2.6 ALP染色检测成骨诱导分化能力 收集各组第三代骨髓间充质干细胞,细胞计数后以3×105/孔接种至6孔板中,培养细胞汇合至60%时更换为细胞成骨分化培养基分别培养7天,每3日换液一次。7d后倒置相差显微镜下观察BMSCs复层生长并出现圆形结节,弃培养液并冲洗,多聚甲醛固定细胞20min后,应用BCIP/NBT底物显色试剂盒,染色20min后流水冲洗,倒置显微镜下拍照。
1.2.7 实时定量PCR检测 Trizol提取各组第3代BMSCs常规培养和成骨诱导7d的RNA,应用微量紫外分光光度计测定RNA的浓度和纯度。应用PrimeScript RT reagent Kit进行cDNA合成,按照使用说明在20μL反转录体系中进行,总反应体系为:上游引物0.5μL,下游引物0.5μL,qPCR SuperMix 10μL,cDNA 1μL,ddH2O 8μL。参照NCBI GenBank数据库,采用Primer primer 5.0软件设计目的基因上下游引物,由华大基因公司合成引物。采用CFX96实时定量PCR仪进行检测,PCR扩增反应条件:95℃3min;95℃10s,55.5℃30s,35个循环。

表1 目的基因的引物序列
1.2.8 统计学分析 各个试验重复三次,定量结果数据以)表示。应用SPSS 17.0统计软件采用t检验和单因素方差分析对试验结果数据进行分析,检验水准为双侧α=0.05。
2.结果
2.1 2型糖尿病大鼠模型建模的确定 6至14周,GK组大鼠体重增长较Wistar组缓慢,且有随着生长周龄增加两组大鼠体重差异逐渐扩大的趋势(图1A)。14周龄的GK组大鼠有多饮、多尿的表现,GK组大鼠的空腹血糖值14.61±5.31显著高于Wistar组的空腹血糖值6.24±0.54(图1B)。GK组大鼠内眦静脉血的空腹胰岛素含量值0.81±0.14显著高于Wistar组(图1C)。OGTT口服葡萄糖耐量试验结果显示GK组大鼠血糖波动幅度较Wistar组大,60min的血糖值达到最高值,120min后Wistar组大鼠血糖值6.4±0.3降至约初始水平,GK组大鼠血糖值22.3±2.1则较初始水平显著上升(图1D)。所有GK组大鼠生理指标都满足空腹血糖值≥11.1mmol/L或OGTT实验120min后血糖大于16.7mmol/L的标准,均建模成功。
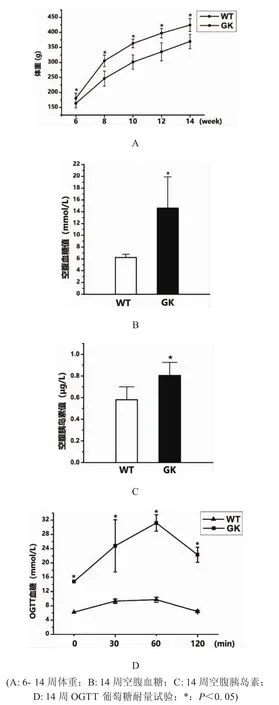
图1 大鼠糖尿病生化指标检测试验
2.2 2型糖尿病对胫骨骨形态计量分析的影响显微CT扫描两组大鼠胫骨形态计量分析的结果见(表2)。GK糖尿病组大鼠胫骨长度显著低于Wistar对照组,结构模型指数SMI显著高于Wistar对照组,而骨矿物质密度BMD、骨小梁厚度Tb.Th、骨小梁间距Tb.Sp及骨体积分数BV/TV在两组间均无明显差异。虽然GK糖尿病组结构模型指数SMI值显著增加显示可能有骨质疏松倾向,但综合骨矿物质密度BMD、骨小梁厚度Tb.Th、骨小梁间距Tb.Sp及骨体积分数BV/TV数值说明2型糖尿病尚未显著引起骨质疏松样改变。
表2 大鼠胫骨显微CT骨形态计量分析,n=8)

表2 大鼠胫骨显微CT骨形态计量分析,n=8)
*:P<0.05
?
2.3 2型糖尿病对BMSCs增殖和凋亡活性的影响 与对照组Wistar BMSCs流式细胞周期结果比较,GK组BMSCs在2型糖尿病微环境中,G1期细胞百分数93.38±0.69%显著高于Wistar组86.72±1.64%,S期细胞百分数2.60±0.46%,G2期细胞百分数4.05±0.31%,都显著低于对照组,表明2型糖尿病微环境对GK组BMSCs具有G1期阻滞作用(图2A,表3)。应用流式细胞仪检测两组BMSCs细胞凋亡,两组BMSCs均只有少量细胞凋亡,GK组BMSCs早期凋亡2.01±0.61%,晚期凋亡11.74±1.47%,凋亡率显著比Wistar对照组高(图2B,表4)。
CCK-8试验结果显示,两组BMSCs在第一天和第二天吸光度值未见明显差异,但第三天开始Wistar组吸光度值显著高于GK组(图3)。GK组细胞生长曲线较平缓,从第三天开始Wistar组BMSCs细胞逐渐步入对数生长期,且扩增能力显著强于GK组BMSCs。
结合增殖和凋亡试验表明,2型糖尿病微环境对BMSCs具有抑制增殖活性、促进细胞凋亡的作用。

图2A 流式细胞仪检测BMSCs细胞周期
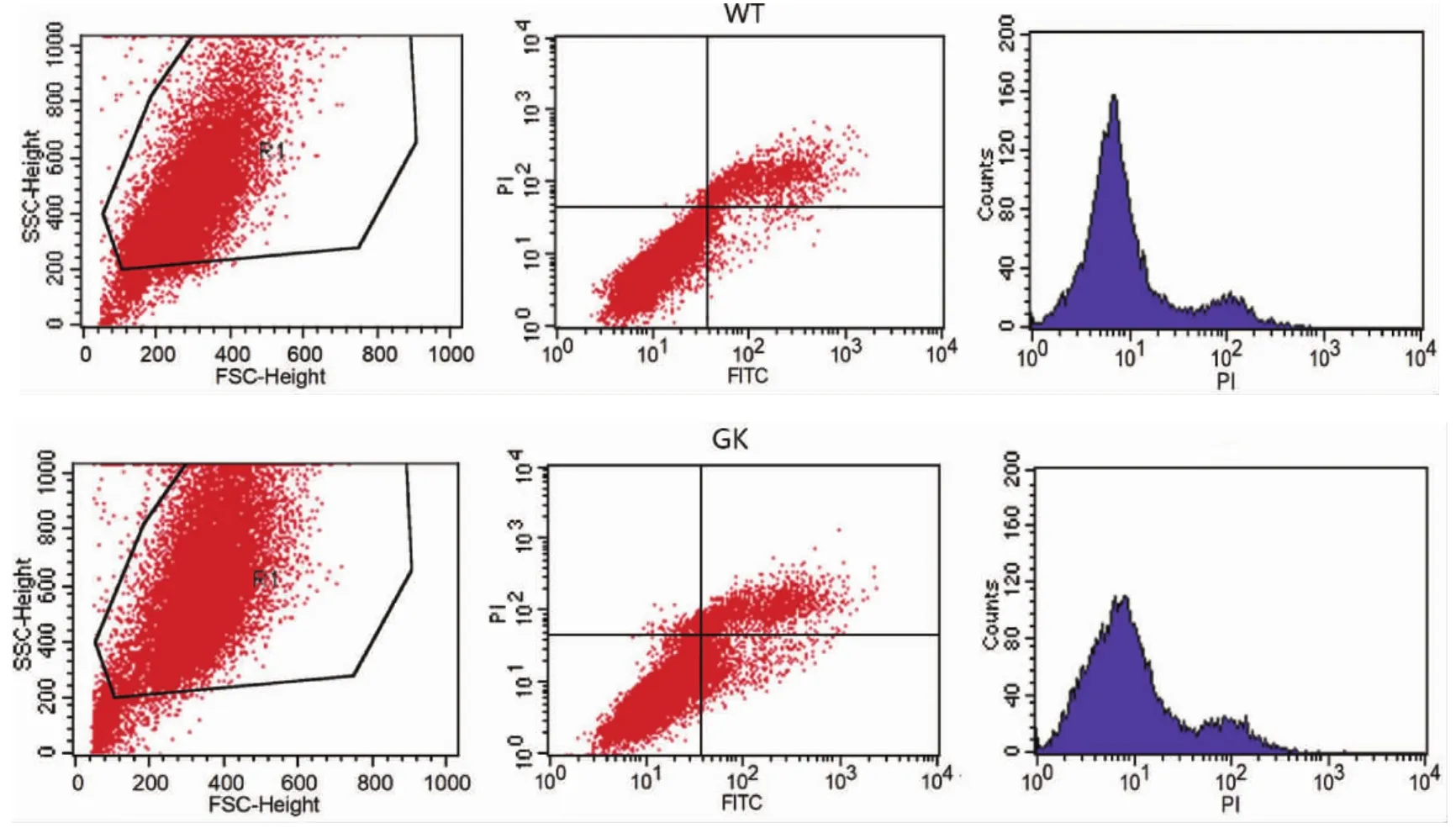
图2B 流式细胞仪检测BMSCs细胞凋亡
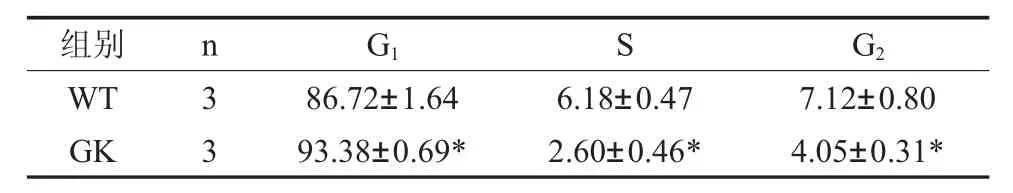
表3 2型糖尿病对BMSCs细胞周期的影响(%)

表4 2型糖尿病对BMSCs细胞凋亡的影响(%)
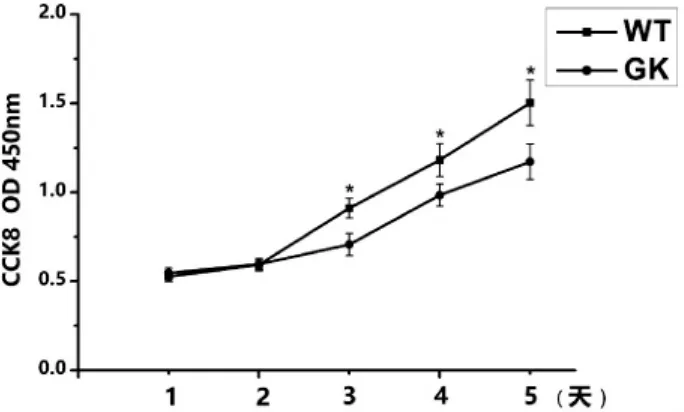
图3 CCK-8试验(*:P<0.05)
2.4 2型糖尿病抑制BMSCs成骨分化能力 两组BMSCs经过7d成骨诱导后,镜下可见GK组和Wistar组BMSCs都形成众多圆形结节,Wistar组圆形结节更大、数更多量。ALP染色结果表明,6孔板中Wistar组较GK组染色更深、着色密度更大。镜下见ALP染色呈蓝色,围绕圆形结节区域,呈条状或团状,GK组染色区域较稀疏(图4-图5)。
两组BMSCs经过7d成骨诱导后,采用qRTPCR对GK组和Wistar组BMSCs成骨分化诱导前后的早期及晚期成骨标志基因表达进行检测,结果发现:成骨诱导前,GK组BMSCs的成骨基因OPN、ALP、Runx2 mRNA表达量(0.14±0.04,0.45±0.13,0.50±0.12)低于 Wistar组(0.29±0.04,0.70±0.08,0.78±0.16);成骨诱导后,GK组BMSCs的成骨基因OPN、ALP、Runx2mRNA表达量(0.34±0.08,0.64±0.11,0.83±0.08)都较成骨诱导前显著增加,但都低于Wistar组(0.82±0.10,1.47±0.06,1.21±0.04),二者差异具有统计学意义。晚期成骨标志物OSX虽然在成骨诱导前两组间无明显差异,但成骨诱导后GK组OSX mRNA表达量0.68±0.05显著低于Wistar对照组0.88±0.04(图 6)。
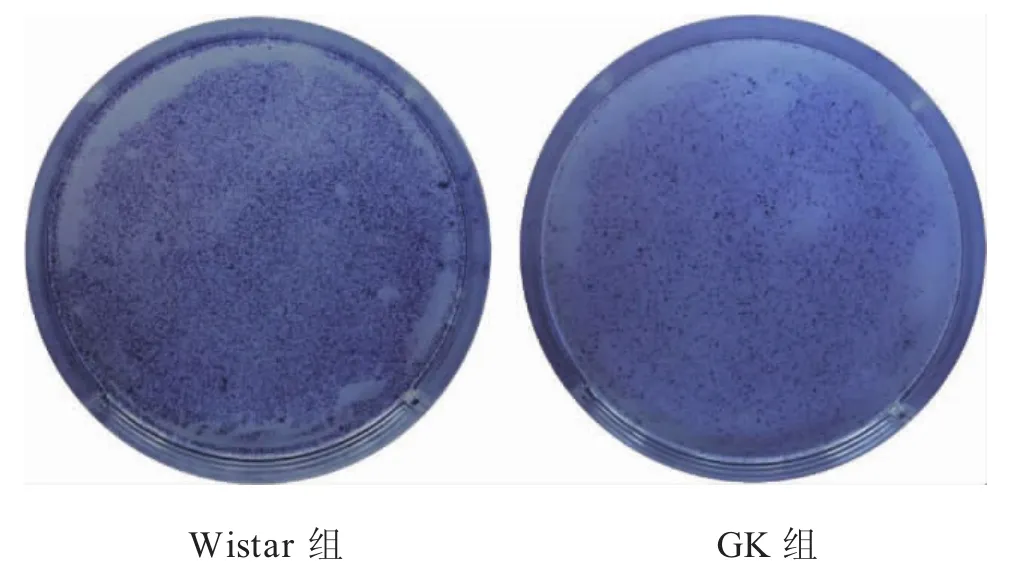
图4 BMSCs ALP染色平皿肉眼观

图5 BMSCs ALP染色倒置显微镜下观

图6 BMSCs成骨标志基因表达(OS:成骨诱导;*:P<0.05)
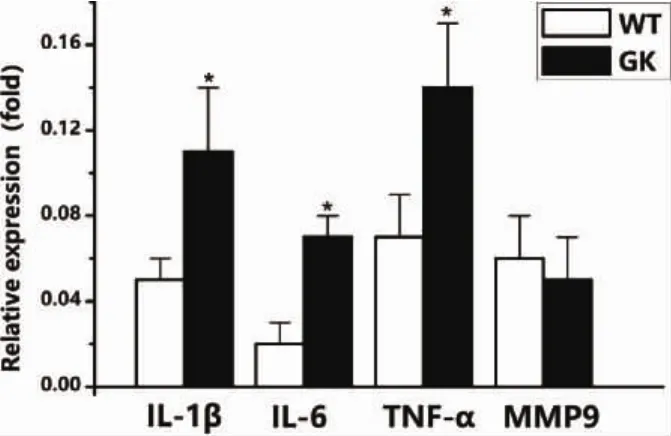
图7 BMSCs促炎因子基因表达(*:P<0.05)
该定性和定量的结果提示,2型糖尿病微环境下调BMSCs早期及晚期成骨标志基因的表达,抑制BMSCs的成骨分化能力。
2.5 2 型糖尿病对 IL-1β、IL-6、TNF-α、MMP9促炎因子表达的影响 应用qRT-PCR对GK组和Wistar组BMSCs的促炎因子基因表达进行检测,结果发现:GK组BMSCs的成骨基因IL-1β、IL-6、TNF-αmRNA 表达量(0.11±0.03,0.07±0.01,0.14±0.03)高于 Wistar组(0.05±0.01,0.02±0.01,0.07±0.02),而 MMP9 mRNA表达量在两组间无显著差异(图7)。IL-1β、IL-6及TNF-α与炎症的发生及进展关系密切,该结果说明2型糖尿病上调BMSCs部分促炎因子的表达,可能引起BMSCs炎症微环境从而影响其成骨活性。
2.6 2型糖尿病对caspase-3、caspase-8凋亡因子表达的影响 应用qRT-PCR对GK组和Wistar组BMSCs的凋亡因子基因表达进行检测,结果发现:GK组BMSCs的凋亡因子caspase-3、caspase-8 mRNA表达量在GK组显著增高,表达量分别为0.27±0.03、0.18±0.03,高于Wistar对照组0.18±0.04、0.14±0.01,P<0.05。该结果与之前流式细胞仪检测的细胞凋亡趋势一致,表明2型糖尿病促进BMSCs凋亡活性。
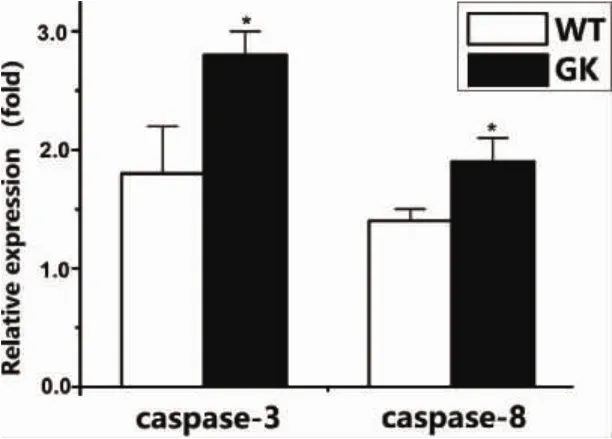
图7 BMSCs凋亡因子caspase3、caspase8基因表达(*:P<0.05)
3.讨论
1型糖尿病和2型糖尿病均增高骨折风险、干扰骨形成及损伤骨愈合。1型糖尿病为胰岛素依赖型糖尿病,在1型糖尿病患者和动物模型中,骨矿物质密度(bone mineral density,BMD)指数降低,骨小梁数量减少,骨髓脂肪组织增多[6]。1型糖尿病大鼠的高糖状态使得Nfic等成骨相关基因表达下降,成骨标记产物降低[7]。临床研究发现1型糖尿病使腰椎骨折风险增高1.3-2.3倍,股骨颈部骨折风险增高约2.6倍,桡骨远端骨折风险增高约1.8倍[8]。2型糖尿病为非胰岛素依赖型糖尿病,大多研究也认为2型糖尿病增加骨折风险。而有研究表明2型糖尿病影响骨代谢活动与时间相关,初始阶段的胰岛素抵抗正性影响骨组织,而长期进展的2型糖尿病则损伤骨代谢活动。也有临床研究发现新确诊的2型糖尿病骨折风险与正常人群无明显差异,而有5年及以上病史2型糖尿病人群的骨折风险显著上升[9-11]。
本研究发现2型糖尿病大鼠模型骨矿物质密度BMD指数与对照组无显著差异,这与之前部分研究结论一致,但也有学者指出BMD指数并未能完全反映骨量变化。通过计算机断层扫描研究2型糖尿病患者髋关节和脊柱,证明2型糖尿病患者具有相似的小梁容积密度,而骨量和横截面积显著降低[12,13]。本研究通过显微CT扫描结果证实2型糖尿病GK大鼠模型的胫骨长度降低、结构模型指数增高。而理论上发生骨质疏松时,骨小梁从板状向杆状转变,结构模型指数增加、骨小梁厚度值减少、骨小梁间距值增加,更多本研究其他的数据包括BMD、骨小梁厚度、骨小梁间距及骨体积分数数值并无显著差异,说明14周内2型糖尿病尚未显著引起骨质疏松样改变。值得一提的是,Burghard等[14]学者发现在骨小梁区域,部分高骨密度面积补偿了2型糖尿病受试者中较低的骨密度面积受试者,从而维持骨小梁整体正常的压缩骨强度;相反,在骨皮质区域,骨骼面积较小,受试者未被高骨密度面积补偿,导致骨质下降、弯曲强度和皮质骨脆性增加,这些发现可能有助于解释2型糖尿病患者髋关节和其他易骨折部位骨折风险增高的现象。有学者[15]报告1型糖尿病患者骨量减少和骨质疏松发病率为48%~72%,2型糖尿病骨质疏松发病率可达20%~60%。糖尿病相关的细胞因子、生长因子、信号通路在问充质干细胞的分化过程中发挥着重要的调控作用,共同导致糖尿病性骨质疏松的发生。由于本研究可能构建2型糖尿病GK模型时间较短,骨计量形态学参数是否会随着糖尿病进展而发生更明显的变化,未来需要进一步探究。
骨髓间充质干细胞一直是骨再生医学的热点,其具备良好的增殖能力、多向分化能力,是理想的再生医学种子细胞,对于骨形成活动及更新具有重要意义。本研究结果表明2型糖尿病促进BMSCs凋亡活性、抑制BMSCs的增殖能力和成骨分化能力,抑制骨形成代谢活动。同时,2型糖尿病上调促炎因子白介素 1β(interleukin-1β,IL-1β)、白介素6(interleukin 6,IL-6)、肿瘤坏死因子α(tumor necrosis factor α,TNF-α)的表达,表明 2 型糖尿病引起BMSCs炎症微环境。有学者[16]研究也发现2型糖尿病大鼠颌骨来源的BMSCs的成骨分化能力受到损害。临床研究[17]报告2型糖尿病患者血样品中骨钙素(osteocalcin,OCN)含量降低,且与IL-6及C反应蛋白含量呈负相关。也有研究表明患有牙周炎的人群也具有显着增加的TNF-α、IL-1β和IL-6水平[18,19],炎症延长并伴有脂质过氧化增加[20]。Pacios等[21]学者指出2型糖尿病使成骨细胞OCN表达降低,肿瘤坏死因子水平增加,而使用肿瘤坏死因子抑制剂可以部分恢复成骨相关因子的表达。肿瘤坏死因子还限制下调其他炎症因子表达的能力,增加核因子-κB活性,从而激活核因子 κB(nuclear factor-kappa B,NF-κB)信号通路,导致成骨细胞fra-1和Runx2基因表达减少,抑制成骨分化能力[22,23]。研究发现2型糖尿病也增加caspase-3凋亡相关基因的表达,促进细胞凋亡的发生[24],本研究结果也验证了这一趋势,但炎症介质影响骨髓间充质干细胞的增殖分化更多具体机制尚需进一步探究。
综上所述,本研究发现2型糖尿病GK大鼠模型短期内未使长骨发生显著骨质疏松样改变;2型糖尿病抑制骨髓间充质干细胞增殖活性及成骨分化能力,上调骨髓间充质干细胞部分促炎因子和凋亡因子的表达。该结果提示,2型糖尿病引起的炎症微环境可能是抑制骨形成代谢的原因之一,研究抑制或逆转炎性介质作用或成为调控恢复2型糖尿病微环境下的骨髓间充质干细胞成骨活性的新思路。
