锑烯吸附金属Li原子的密度泛函研究*
2019-03-11栾晓玮孙建平王凡嵩韦慧兰胡艺凡
栾晓玮 孙建平 王凡嵩 韦慧兰 胡艺凡
(华北电力大学电气与电子工程学院, 北京 102206)
(2018 年9 月3日收到; 2018 年11 月23日收到修改稿)
锑烯(antimonene)是继石墨烯和磷烯之后出现的新型二维材料, 在锂离子电池等领域受到关注. 本文基于第一性原理的密度泛函理论, 计算研究了锑烯对Li原子的吸附特性, 包括Li原子的最稳定吸附构型、吸附密度以及吸附Li原子的扩散路径. 结果表明: Li原子最稳定的吸附位置位于谷位, 即底层Sb原子之上、顶层三个Sb原子中心位置, 吸附能为1.69 eV, 吸附距离为2.81 Å; 能带计算发现, 锑烯为带隙宽度1.08 eV的间接带隙半导体, 吸附Li原子后费米能级上升进入导带, 呈现出金属性; 原子分波态密度分析发现, Sb原子的p电子态和Li原子的p和s电子态形成明显的共振交叠, 表现出杂化成键的特征; 随着吸附Li原子数量增加, 锑烯晶格结构和电子结构发生较大变化. 通过微动弹性带方法计算发现, Li原子在锑烯表面的扩散势垒为0.07 eV,较小的势垒高度有利于快速充放电过程.
1 引 言
锂离子电池具有高能量密度、高存储能量、循环寿命长和低损耗的优点, 在日常电子产品、储能系统中得到广泛应用. 随着锂离子电池在电动汽车、储能电池、航空航天以及军事等相关领域需求的快速增长, 人们对锂离子电池的可逆性容量、倍率充放电能力和循环稳定性等性能提出了更高的要求[1,2]. 由于锂离子电池性能很大程度上受到电极材料储锂容量和循环速率的制约, 传统负极材料的储锂性能已经接近理论极限. 以石墨烯和磷烯为代表的新型二维材料具有独特结构和优异的电学性能, 在锂离子电池负极材料领域具有很好的应用前景, 引起了人们的极大关注. 2004年, 英国科学家Geim[3]采用机械剥离法, 首次获得能够在外界条件下稳定存在的石墨烯. 石墨烯是碳的一种同素异形体, 是碳原子以sp2杂化形成的六角蜂窝状单层薄膜, 层厚约为0.335 nm. 石墨烯具有优异的导电性、较高的机械强度等优点. 由于石墨烯独特的单层碳原子结构, 使Li离子不仅可以存储在石墨烯片层的两侧, 还可以存储在石墨烯片层的边缘和中间, 其最佳吸附位置是碳六边形的中心[4]. 其理论比容量为740—780 mA·h/g, 锂离子容易在其中嵌入和脱嵌, 使得电极电导率较高, 表明石墨烯作为锂离子电池负极材料具有其优越性[5,6]. 有研究表明, 在石墨烯上掺杂其他元素如硼、氮等元素可以改变石墨烯的电学性能, 且氮掺杂石墨烯作为锂离子电池负极材料具有稳定的电化学性能和更大的储锂能力[7-9]. 磷烯是最新出现的二维半导体材料, 且单层和少层黑磷具有直接带隙和高的电子迁移率, 其直接带隙的特性弥补了石墨烯用作半导体器件的不足, 在微电子器件方面有很好的应用前景[10].在锂离子电池领域, 实验研究表明, 磷烯包覆的锂离子负极材料可以提升电池的库仑效率、容量以及循环稳定性[11,12]. 磷烯放电电位范围为0.4—1.2 V,锂离子可以在其中快速定向扩散, 在0.2 C电流中放电, 其比容量达到2786 mA·h/g. 理论计算表明,磷烯带隙为0.91 eV, 导电性好, 且具有较高的吸附能和较低的扩散势垒, 其中沿锯齿形方向的单层磷烯扩散势垒仅为0.09 eV, 表明其具备快速充放电的潜力[14], 高于基于石墨烯的锂离子电池负极材料[13-15]. 此外, 吸附过渡金属元素可以提高磷烯作为锂离子电池负极材料的储锂性能[16]. 但是磷烯直接应用于各种器件较为困难, 主要原因是磷烯在空气中极易氧化, 结构可在数小时内被破坏. 通过修饰、掺杂、涂覆与复合等方式钝化和功能化磷烯,可以抑制磷烯的氧化, 在一定程度上提高稳定性并改善性能[17].
继石墨烯和磷烯之后, 最近出现的二维材料锑烯(antimonene), 由于具有良好的化学稳定性和独特的物理特性, 在半导体电子器件、锂离子电池等领域引起广泛关注[18,19]. 它是一种完全由Sb原子组成的具有皱褶起伏的类石墨烯结构, 是一种新型二维间接带隙半导体材料, 具有优良的光电子学和自旋电子学性质[20,21]. 目前, 有报道实验上已经可以通过机械剥离、液相剥离、气相生长等方法制备化学稳定的锑烯[22]. 在锂离子电池领域, 人们之前对Sb块体的研究发现, 其理论嵌锂容量可达到660 mA·h/g, 嵌锂电位为0.8 V, 比金属锂稍高的电位能够有效避免枝晶的出现, 提高电池安全性.实验发现, 块体锑材料在嵌脱锂时存在较大的体积膨胀和收缩, 导致电池循环特性变差, 缩短了电池工作寿命. 对于多层和单层锑烯, 随着厚度减小,由于量子尺寸效应, 其电子结构由金属型向半导体转变, 会对锂原子吸附特性产生重要影响; 此外,随着尺寸和维度减小, 体积效应会逐渐减弱, 采用锑烯电极材料的电池循环寿命可望得到延长, 比表面积增大, 也有利于提高储锂容量. 因此, 开展锑烯的理论和实验研究具有重要意义. 理论计算方面, 有报道锑烯对于H2O, O2等小分子有较弱的物理吸附, 而对于H, C, N, O, Al等原子吸附较强[23]. 此外, 结构缺陷、衬底和吸附都会对锑烯稳定性和电子结构带来影响[24-26]. 但是, 目前关于锑烯吸附Li原子的系统研究工作还少见报道. 本文用密度泛函理论计算方法系统研究了锑烯对Li原子的吸附特性, 其中包括Li原子的最稳定吸附构型、吸附密度以及Li原子的扩散路径, 揭示出锑烯具有作为锂离子电池负极材料的良好潜力, 这一结果可以为实验研究提供借鉴. 需要指出的是, 实际应用中, 由于制备难度和团聚效应, 直接采用单层锑烯作为锂离子电池电极材料存在困难, 更大可能是采用多层锑烯材料. 多层结构当层间距较大时, 嵌入其间的锂原子作用类似单层吸附; 但当层间距变小时, 加之堆叠方式可能不同, 结构会比较复杂, 需要大量详细的分析计算, 对多层结构有待进一步研究.
2 计算的细节
文中的计算是采用基于密度泛函理论的VASP软件包来进行的. 其中, 能量交换关联能函数采用Perdew-Burke-Ernzerhof (PBE)形式的广义梯度近似 (general gradient approximate)法, 价电子与离子之间的相互作用势采用缀加平面波赝势法(pseudopotential augmented wave)来描述. 结构优化和计算时采用截断能为500 eV的平面波基组展开, 在布里渊区积分计算时采用了VASP软件推荐的原点在Γ点的Monkhorst-Pack型网格,k点取值为11 × 11 × 1. 弛豫计算时核离子步收敛精度取为0.01 eV/Å, 能量收敛精度取为1 ×10-4eV. 选用4 × 4 × 1锑烯原胞共32个原子,Z方向上真空层取20 Å厚度以避免层间干扰, 并考虑自旋极化作用. 结构优化时, 对所有原子进行弛豫. 原子的吸附能定义为:Ead=Eantimonene+Eatom-Eadsorb, 式中Eatom为单个Li原子的能量,Eantimonene为本征锑烯的能量,Eadsorb为吸附之后整个体系的能量. 根据这个定义,Ead值越大表示吸附能越大, 吸附越稳定.
3 结果与分析
3.1 本征锑烯
为了和吸附Li原子之后的锑烯做对比, 首先对本征锑烯进行计算. 选取4 × 4本征锑烯超晶胞, 如图1(a)和图1(b)所示, 褐色圆点表示Sb原子, 可以看出Sb原子分为上下两层, 呈六边形周期性分布, 为使立体效果更好, 将俯视图稍做倾斜.本征锑烯中近邻Sb原子间距为2.87 Å.
图1(c)是4 × 4本征锑烯取费米能级EF= 0 eV时的能带图, 从图1(c)可以看出, 锑烯能带中价带顶起伏大、色散较明显, 导带底变化较平缓. 能带图中, 价带顶落在布里渊区中心G点, 导带底在G和K点之间, 计算得到的带隙宽度为1.08 eV,为间接带隙半导体. 用PBE方法计算存在带隙偏小的问题, 实际锑烯的带隙会更大. 图1(d)是4 ×4本征锑烯态密度图, 可以看出存在明显的带隙,而且态密度分布相对于费米能级不对称. 以上结果与文献报道一致[23], 符合一般认识, 表明我们选择的计算方法和参数设置是可靠的.
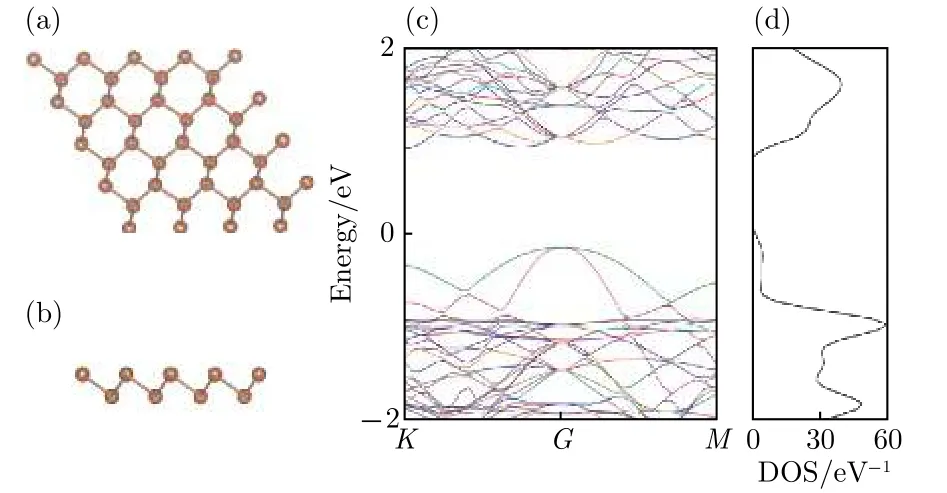
图1 本征锑烯 (a) 俯视图; (b) 侧视图; (c) 能带图;(d) 态密度图Fig.1. Pristine antimonene: (a) The top view; (b) the side view; (c) the band structure; (d) the density of states.
3.2 锑烯吸附单个Li原子
为了研究单个Li原子在锑烯上最稳定的吸附构型, 选取4 × 4本征锑烯超晶胞, 吸附一个Li原子, 考虑三种可能的吸附位置: 顶位, 在顶层Sb原子正上方; 谷位, 在顶层三个Sb原子中心, 底层一个Sb原子上方; 桥位, 在顶层两个Sb原子联线中心. 先结构优化, 对体系的各个原子进行弛豫. 结果发现桥位上的锂原子在优化之后回到了谷位的位置, 所以下面我们就谷位和顶位两种结构进行分析. 两种初始构型和优化构型分别如图2、图3所示, 结构图中褐色圆点表示Sb原子, 绿色圆点表示Li原子, 为使立体效果更好, 将俯视图稍微倾斜.

图2 (a)和(c)分别为谷位吸附结构初始构型的俯视图和侧视图; (b)和(d)分别为其优化构型的俯视图和侧视图Fig.2. The antimonene structure of Li adsorbed on vacancy site: (a) The top view before optimization; (b) the top view after optimization; (c) the side view before optimization; (d) the side view after optimization.
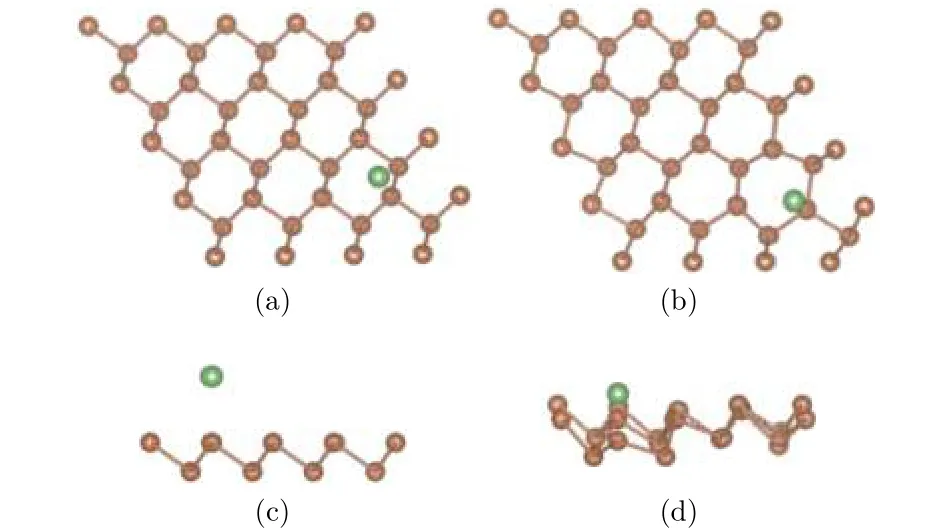
图3 (a) 和 (c) 分别为顶位吸附结构初始构型的俯视图和侧视图; (b) 和 (d) 分别为其优化构型的俯视图和侧视图Fig.3. The antimonene structure of Li adsorbed on top site: (a) The top view before optimization; (b) the top view after optimization; (c) the side view before optimization;(d) the side view after optimization.
通过对比两种结构的初始构型和优化结果, 可以发现Li原子趋向于吸附在谷位, 也即三个Sb原子形成的中心位置. 从两种结构的俯视图来看, 顶位和谷位初始构型的Li原子经过优化后, 位置没有明显改变. 而从两种结构的侧视图来看, 顶位的Li原子相较于初始结构的高度降低, 谷位的Li原子高度相对变化较小.
表1给出了两种构型的吸附构型、Li原子的吸附能、吸附距离和电荷转移量. 其中吸附能Ead定义为是Li原子吸附锑烯之后体系的能量,Eantimonene是本征锑烯能量,Eatom是Li原子能量. 可以看出,Li原子吸附的吸附能适中, 属于化学吸附, 其在顶位的吸附能是1.67 eV, 谷位的吸附能是1.69 eV.吸附能越大, 通常表示吸附后的结构越稳定, 由谷位的吸附能最大可知, 谷位吸附结构最稳定. 吸附稳定性首先可以从吸附原子成键的角度进行分析,从成键时配位原子的数量来讲, 谷位的Li原子可以与周围三个Sb原子以及下层Sb原子发生相互作用成键. 而顶位的Li原子只和一个Sb原子进行相互作用成键. 其次, 从优化结构中Li原子和最近邻的Sb原子的距离可以看出, 谷位吸附距离最短,为 2.81 Å, 顶位吸附距离稍长, 为 2.83 Å, 成键距离短通常表示相互作用强, 说明谷位Li原子吸附最稳定. 从Barder电荷转移也可以进行分析. 由表1可以看出, 吸附过程中, Li原子几乎完全失去1个价电子, 而邻近的Sb原子得到电子, 特别是吸附Li原子最近邻的Sb原子得到较多的电荷. 谷位吸附结构中, 与Li原子最近邻的Sb原子得到的电荷最多, 为0.32e, 其次是顶位结构, 0.31e, 间接表明了谷位结构中Li原子和Sb原子的相互作用较强.

表1 锑烯吸附Li原子的两种结构的特性Table 1. Properties of two Li adsorbed antimonene configurations.
由于谷位吸附构型对应于最稳定构型, 所以针对这种构型进行进一步的分析计算. 计算得到的谷位吸附Li原子的锑烯的能带、总态密度(DOS)和原子分波态密度(PDOS)如图4所示.
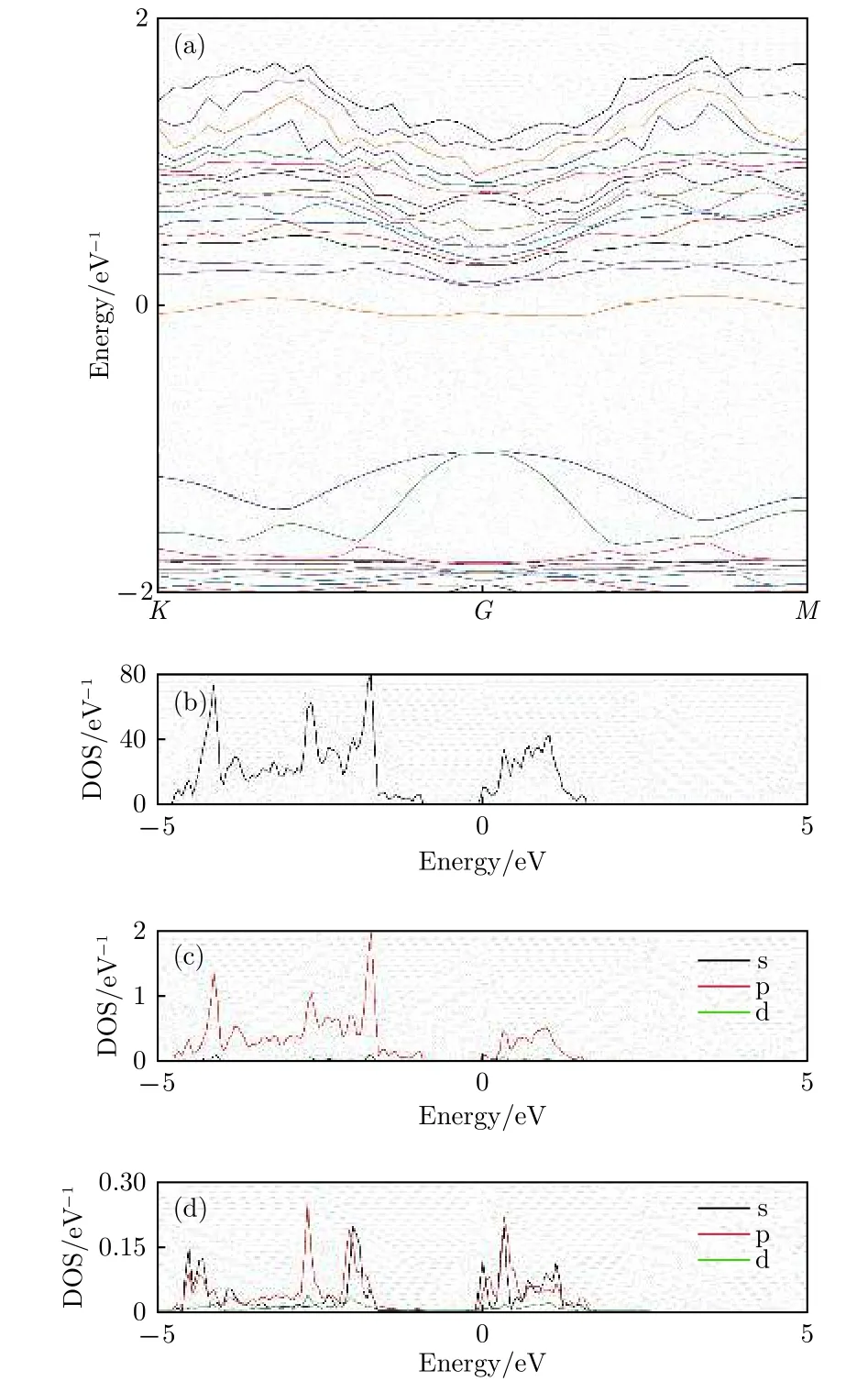
图4 谷位吸附构型 (a)能带图; (b)总态密度;(c) Sb分波态密度; (d) Li分波态密度Fig.4. The band structure and density of Li adsorbed antimonene: (a) The band structure; (b) the total density of states; (c) the partial density of states of Sb atom;(d) the partial density of states view of Li atom.
从图4(a)与本征锑烯相比较能带有明显的改变. 计算得到的谷位吸附结构费米能级在-1.64 eV处, 比本征费米能级有所升高, 以费米能级为零点做图, 可以看出在EF= 0 eV附近, 带隙间靠近导带底附近出现了杂质能带, 而费米能级穿过杂质能带, 呈现出N掺杂特征. 相比本征锑烯, 吸附后能带在价带顶附近变化不大, 而在导带底附近分布更密, 起伏更大. 从谷位吸附Li原子的锑烯总态密度也可以清楚看出, 相比于本征锑烯, 费米能级EF=0 eV上移进入导带. 更进一步, 从Sb原子和Li原子的分波态密度可以看出, 总体上, 主要是Sb原子的p电子态和Li原子的s, p电子态在-5—2 eV区间发生杂化交叠, 产生出一系列共振峰, 表现出Li和Sb原子杂化成键的特征. 具体而言, 费米能级处的杂质能带是由Sb原子的p电子和Li原子的s, p电子交叠形成, 而价带顶处由于没有共振峰, 所以能带不受影响, 这和前面能带分析的结果一致. 由于费米能级处能带变化较大, 对体系导电特性的影响显著, 而导电性的增强对于锂离子电池电极材料而言显然是有利的, 有利于减少充放电时的损耗.
图5给出了顶位吸附Li原子时, 锑烯的总态密度和分波态密度. 从图5可以看出, 两种吸附构型对应的总态密度图相似, 杂化峰位置基本没变,但相对高度发生了变化; 谷位吸附构型相对于顶位吸附, 在-1—5 eV能量区间, 杂化峰高度有一定程度的提高, 显示Sb原子和Li原子相互作用增强. 从Sb的PDOS图中可以看出, 这一变化主要来源于Sb的p电子轨道贡献; 另外, 对比两种构型Li原子PDOS中p轨道和s轨道对杂化峰的贡献, 发现谷位吸附时p轨道在杂化轨道所占比重上升, s轨道相对下降, 这反映出谷位吸附时Li原子和多个Sb原子成键的状态. 态密度分析结果和前面对吸附能、吸附距离和bader电荷分析得到的结果一致.

图5 顶位吸附结构态密度图 (a)总态密度; (b) Sb分波态密度; (c) Li分波态密度Fig.5. The density of states of the Li adsorbed structure:(a) The total density of states; (b) the partial density of states of Sb atom; (c) the partial density of states view of Li atom.
图6给出了Li原子吸附后, 反映吸附结构中各个原子电荷转移状况的差分电荷密度图. 结构图中褐色圆点表示Sb原子, 绿色圆点表示Li原子,黄色和蓝色表示电荷密度高低. 由图6可以看出,总体上谷位吸附结构中Li原子是失去电子的状态,而Li原子所失去的电荷不但转移到了临近的Sb原子之上, 而且更远处的Sb原子上也发生了电荷的相互转移. 综合前面对态密度的分析, 可以发现, Li原子吸附成键, 类似于金属键, 具有非局域的特征.
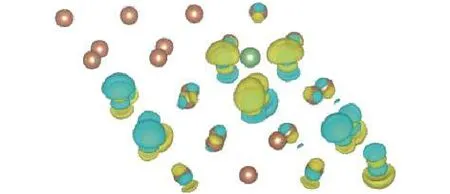
图6 谷位差分电荷密度Fig.6. The differential charge density of the Li adsorbed structure.
3.3 锑烯吸附多个Li原子
为研究吸附Li原子的密度对整个锑烯构型的影响, 分别构建锑烯吸附3个、6个和9个Li原子的吸附模型, 吸附位置都选择最稳定的谷位. 图7、图8分别是吸附3个和9个Li原子的模型图, 其中褐色圆点表示Sb原子, 绿色圆点表示Li原子,采用自动优化的方法对所有原子进行弛豫.
由Li原子不同吸附密度的弛豫结果可以看出,锑烯吸附Li原子之后, 由于和Sb原子间的相互作用, 导致自身原子位置发生偏移. 图7中, 从吸附3个Li原子的结果可以看出, Li原子在水平面内,相对于谷位位置略有偏移, 和Li原子近邻的Sb原子在水平面内位置变化不大, 底层Sb原子在垂直方向下降, 总体偏移不大. 随着吸附原子数量增多,如图8锑烯吸附9个Li原子所示, Li原子吸附的周期性结构一定程度上遭到破坏, 无论是平面方向还是垂直方向, 结构变化较大, 可以认为出现了结构混乱, 这和Li与Sb原子间相互作用比较强有关.
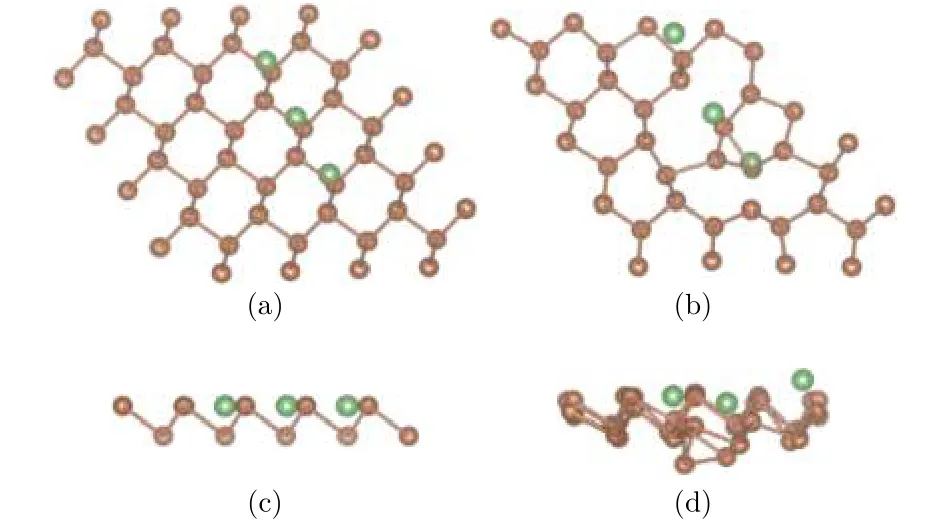
图7 (a)和(c)分别为锑烯吸附3个Li原子结构初始构型的俯视图和侧视图; (b)和(d)分别为其优化构型的俯视图和侧视图Fig.7. The configurations of antimonene adsorbing three lithium atoms: (a) The top view before optimization;(b) the top view after optimization; (c) the side view before optimization; (d) the side view after optimization.
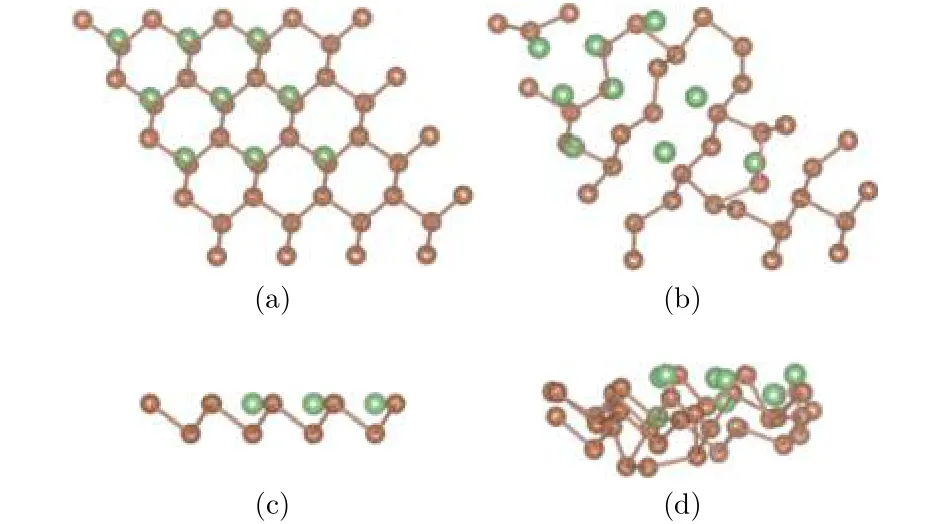
图8 (a)和(c)分别为锑烯吸附九个Li原子结构初始构型的俯视图和侧视图; (b)和(d)分别为其优化构型的俯视图和侧视图Fig.8. The configurations of antimonene adsorbing nine lithium atoms: (a) The top view before optimization;(b) the top view after optimization; (c) the side view before optimization; (d) the side view after optimization.
我们定量计算的不同数量的Li原子吸附构型中, 平均每个Li原子的平均吸附能Ead定义为:其中Eatom为Li原子的能量,n为吸附Li原子的数量. 锑烯吸附Li原子数量为1, 3, 6, 9时,原子平均吸附能分别为1.69, 1.15, 0.94, 0.97 eV. 可以看出,随着锑烯吸附Li原子数量从1个增大到6个, 原子平均吸附能从1.69 eV逐渐下降到0.94 eV, 这符合化学吸附的一般规律. 一般认为, 随着吸附密度增大, 吸附Li原子间的相互作用增强, 会引起Li原子和临近Sb原子间相互作用的削弱. 另一方面,可以看到, 这一变化趋势随着吸附密度的进一步增大而遭到破坏, 当吸附数量达到9个时, 平均吸附能增大到0.97 eV, 而这应该是Li原子吸附引起锑烯结构显著改变引起的.
图9给出了锑烯吸附3个Li原子的态密度,和图5中吸附单个Li原子态密度比较, 可以看出,它们有少量不同之处, 总体差别不大. 相似之处在于, 如图9(a)和图9(b)所示, 吸附体系总态密度和Sb原子分波态密度中峰的位置、形状和相对高度大致未变, 显示体系总体电子结构未变; 差别之处是, 如图9(c)中所示, 单个Li原子吸附时, Li原子分波态密度在-5—2.5 eV能量范围呈现为多个分立的共振峰; 3个Li原子吸附时, 靠近-5 eV低能端的峰消失, 变成连续能带, 这一变化主要是由于Li原子间的相互作用产生的. 总态密度图相差不大, 说明吸附Li原子对锑烯整体电学特性的影响不大.
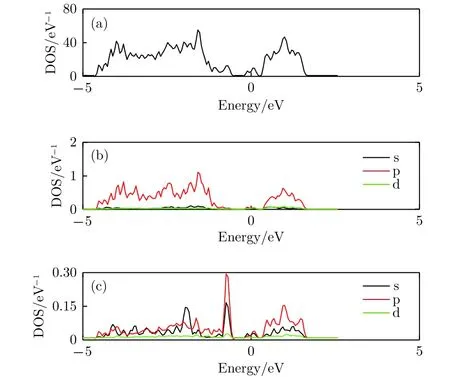
图9 锑烯吸附三个Li原子态密度图 (a) 吸附结构的总态密度图; (b) Sb原子的分波态密度; (c) Li原子的分波态密度Fig.9. The density of states of antimonene adsorbing three lithium atoms: (a) The total density of states of the structure; (b) the partial density of states of Sb atom;(c) the partial density of states view of Li atom.
图10给出了锑烯吸附九个Li原子的态密度图, 和图5相比, 锑烯吸附九个Li原子的态密度图变化较大. 从图10(a)和图10(b)中可以看出, 锑烯的总态密度和Sb原子分波态密度的形状发生了改变, 整体更加平缓. 此外, 带隙变窄几乎消失, 呈现典型的金属性. 由图10(c)也可以看出, 原来Li原子分立的共振峰很大程度上消失, 变成连续分布的能带, 说明随着吸附原子数量的增加, 锑烯电子结构有逐渐从孤立原子吸附状态向合金化转变的趋势. 综合以上分析可以看出, 随着吸附Li原子数量的增加, 一方面对锑烯的晶体结构造成较大扰乱, 另一方面也会引起电子结构的改变. 对于单层锑烯而言, 要达到最大储锂密度, 稳定性应该是需要考虑的重要因素. 实际应用中, 结构更加稳定的多层锑烯模型应该更符合实际状况, 有待下一步研究.

图10 锑烯吸附九个Li原子态密度图 (a) 吸附体系的总态密度图; (b) 体系中Sb原子的分波态密度; (c) 体系中Li原子的分波态密度Fig.10. The density of states of antimonene adsorbing nine lithium atoms: (a) The total density of states of the structure ; (b) the partial density of states of Sb atom;(c) the partial density of states view of Li atom.
3.4 吸附Li原子的迁移
Li原子在锂离子电池电极材料内的扩散难易直接影响电池的充放电速率, 为了弄清楚吸附Li原子在锑烯表面的扩散迁移过程, 即从稳定吸附位置迁移到邻近的另一个稳定位置的路径, 采用VASP自带的微动弹性带(NEB)过渡态搜索的方法进行了计算研究. 首先设定初态和终态两个稳定位置, 然后选取插点数量为三个, 通过计算得到从初态到终态的迁移路径上结构和各个状态的能量变化.
图11结构图中褐色表示Sb原子, 绿色表示Li原子. 图11中绘出的5个Li原子表示了Li原子的扩散迁移路径, 最左和最右的Li原子分别表示初态和终态的稳定吸附位置, 记为状态1和状态5, 中间三个对应于扩散中间状态2, 3, 4, 其中状态3位于扩散路径的中点. 图12给出了迁移过程中从初态到终态体系能量的变化. 可以看出, 状态3能量明显最高, 而且恰好位于路径中点, 根据对称性判断, 状态3应该对应于势能面鞍点, 称为过渡态. 通常规定初态和过渡态的能量差为扩散势垒, 势垒高度决定了扩散发生的难易程度, 势垒低,则从一个稳定态迁移到另一个稳定态越容易, 外加电场条件下迁移发生也越快, 反之则越困难越慢.从图11中数据可以计算得到扩散势垒高度Eb=0.07 eV, 锂离子电池充放电的快慢程度常用倍率特性描述, 大倍率充放电能力在很多场合非常重要. 其中电极材料对锂离子的扩散能力是影响充放电倍率的一个重要因素, 扩散势垒低说明扩散和迁移更加迅速, 较小的势垒高度表明, 锑烯用作电极材料有利于发生快速充放电.
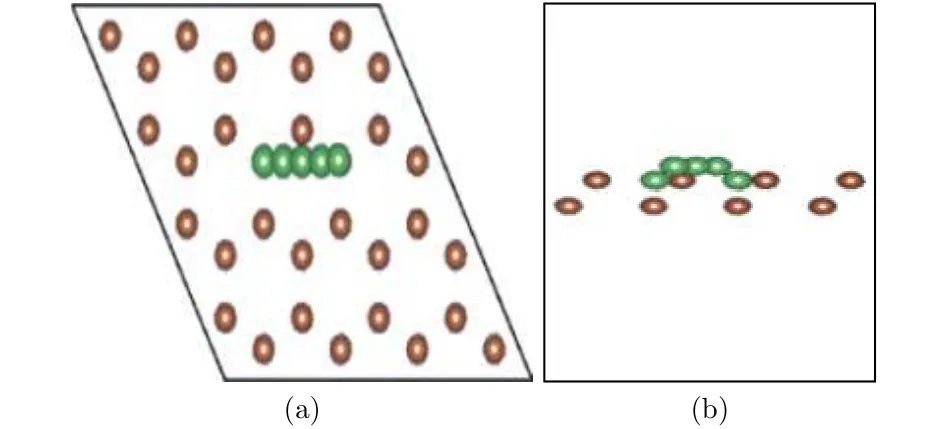
图11 Li原子扩散迁移路径 (a) 俯视图; (b) 侧视图Fig.11. The Li diffusion path on antimonene: (a) The top view; (b) the side view.
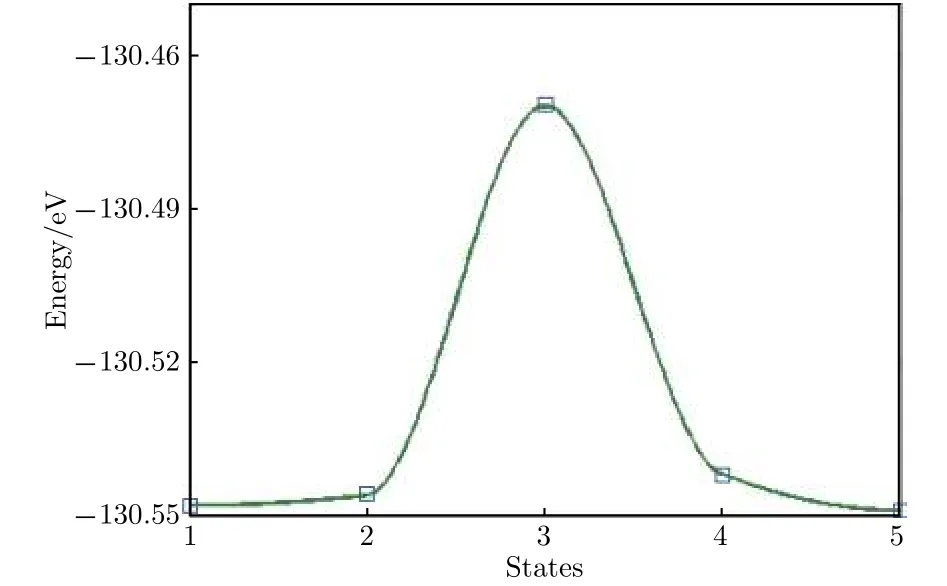
图12 Li原子扩散迁移势垒Fig.12. The diffusion energy barrier of Li atom.
4 结 论
锑烯是继石墨烯和磷烯之后, 实验中最新制备获得的二维半导体材料, 由于具有很好的稳定性和独特的电学特性, 在半导体器件和锂离子电池等领域引起人们的广泛研究兴趣. 本文采用第一性原理的密度泛函方法, 通过理论模拟计算, 系统地研究了Li原子在单层锑烯上的吸附特性; 发现最稳定的吸附构型是谷位, 吸附能为1.69 eV, 吸附距离为2.81 Å. 电子结构计算发现, 锑烯为带隙宽度1.08 eV的间接带隙半导体, 吸附Li原子后费米能级上升穿过杂质能带, 呈现出金属性; 随着吸附Li原子数量的增加, 单层锑烯晶体结构和电子结构都出现了较大改变. 实际应用中, 采用多层锑烯结构或复合结构(如石墨烯/锑烯), 有望提高稳定性从而获得更高的储锂密度; 通过NEB过渡态搜索的计算方法研究Li原子迁移路径, 发现Li原子扩散势垒为0.07 eV. Li原子在锑烯上的迁移势垒较小, 有利于快速充放电过程. 综上所述, 锑烯有作为锂离子电池负极材料的优良潜力, 本文研究结果可为相关实验研究提供理论参考.
