美国FDA的新药临床试验申请和药品审批
2019-03-10谭燕
谭燕
摘 要 美国FDA的“研究新药申请”为新药临床试验申请,新药的审批工作主要由美国FDA的药品审评与研究中心和生物制剂审评与研究中心负责。本文介绍美国FDA新药临床试验申请的审核程序、临床试验的监管方式、药品审评的沟通机制和FDA的审评资源,并提出了对我国临床试验申请审批的借鉴建议。
关键词 美国FDA 新药 临床试验
中图分类号:R951 文献标志码:C 文章编号:1006-1533(2019)03-0064-03
Application of new drug clinical trial and drug approval in FDA
TAN Yan*
(School of Medical Devices, Shanghai University of Medicine & Health Sciences, Shanghai 201318, China)
ABSTRACT Investigational New Drug Application (INDA) in FDA is an application of new drug clinical trials. The approval of new drugs is mainly carried out by the FDAs Center for Drug Evaluation and Research and the Center for Biologics Evaluation and Research. The INDA review process, the management of clinical trials, the communication system of drug review and the review resources were discussed in this article so as to hopefully provide reference for the application and approval of clinical trial in China.
KEy WORDS US FDA; new drug; clinical trail
美國FDA的“研究新药申请”(Investigational New Drug Application, INDA)为新药临床试验申请,新药的审批工作主要由美国FDA的药品审评与研究中心(Center for Drug Evaluation and Research, CDER)和生物制剂审评与研究中心(Center for Biologics Evaluation and Research, CBER)负责。FDA的药品注册管理法规体系按照法案、管理规定、技术指导原则的层级自上而下共同构成[1] 。FDA通过技术指导原则对制药企业的研发过程给予科学的、规范性的指导和建议,同时通过实施药品的生产质量管理规范、非临床研究质量管理规范和临床研究质量管理规范进行监管。在FDA的管理规定中,INDA的申请人可是个人、制药企业、政府机构、学术机构、私人组织或其他组织,目的可是上市销售或仅作为科学研究。INDA作为新药研发过程中的重要探索阶段的临床试验申请,FDA对其申请资料数量的要求是较为灵活的。
1 INDA的审核程序
1.1 审核流程
FDA对INDA采用备案制,申请人在提交申请资料前可申请INDA前咨询,向FDA咨询临床前研究、临床研究和INDA的申请资料等有关问题。FDA于收到首次INDA后的30 d内进行审核。虽然30 d后申请人没有收到任何回复即可开始进行临床试验,但因如FDA认为临床试验存在问题可随时发出暂缓或终止试验的行政指令,故而申请人在未收到FDA的回复前一般不会开展临床试验。
1.2 申请资料
INDA的申请资料主要包括:①INDA申请表;②药品介绍和总体研究计划说明,包括第1年欲进行的临床试验种类、在药品或相关药品的动物毒理学研究数据或以前进行的人体毒理学研究数据的基础上预测的所有重大风险等;③研究人员手册,包括原料药结构式,已知的药品在动物和人体中的药、毒理学作用和药代动力学,已获得的有关药品安全性和有效性的文献资料等;④协议,包括详细的临床研究方案;⑤化学、生产和控制信息,其中每个临床试验阶段都要求提交能确保药品特性和质量的充足的数据,但不同临床试验阶段对数据量的要求不同,Ⅰ期临床试验需提交的数据通常重点放在原料药和相关辅料的鉴定、控制方面;⑥药、毒理学研究数据;⑦已有的药品在人体中的研究结果(文献数据);⑧其他数据,如精神系统疾病用药可能有的依赖性和滥用可能性、放射性药品需提供充足的在动物或人体中的研究数据等。
申请人首次提交INDA后会获得一个INDA号码,此后可随时、主动地多次提交INDA的补充资料。虽然法规给予了需提交资料的数量和程度的很大灵活性,但FDA认为,在满足Ⅰ期临床试验安全性要求的基础上应减少提交资料的数量。若遵照FDA的技术指导原则,申请Ⅰ期临床试验所需提交的资料的厚度通常不超过2 ~ 3 in(1 in=2.54 cm)。
2 临床试验的监管方式
INDA通过审核后,申请人即可开始进行临床试验,而确保临床试验的质量对保障药品的安全性、有效性有着重要作用。FDA对开展药品临床试验的机构并无认证机制,只要临床试验的主要研究者是有行医执照的有资质的医生就可以了。
2.1 行政指令
FDA监管临床试验的行政指令有3种,分别为暂缓、终止和静止,其中暂缓又有完全和部分暂缓之分。无论INDA是尚处于FDA审核阶段、还是已在进行临床试验,只要FDA发现临床试验存在严重不足,均可能发出临床试验暂缓的行政指令,申请人必须暂停临床试验。严重不足包括:受试者有受到不合理的或严重的疾病或伤害的风险;风险评估信息不充分;Ⅱ、Ⅲ期临床试验方案存在缺陷;研究人员手册不完整或有信息不正确;临床试验的研究者不具备应有的资质等。申请人改正了上述不足或满足了FDA的要求后,FDA会批准恢复进行临床试验。申请人也可向FDA提出恢复申请并提交相应资料,FDA将在收到恢复申请后30 d内给予书面答复。
如临床试验在2年或更长时间内没有进行或处于暂缓状态1年以上,FDA将把INDA转为静止状态并通知申请人,申请人需于30 d内向FDA提出恢复申请。
如FDA认为临床试验会对受试者健康产生巨大的直接危害,CDER或CBER可在任何时候通知申请人立即终止临床试验。
2.2 现场检查
1977年,FDA建立了“生物研究监测程序”(Bioresearch Monitoring Program, BiMo)[2],用于对临床试验进行现场检查,以保障受试者安全并确保临床试验数据的质量和真实性,由其监管事务办公室(Office of Regulatory Affairs, ORA)执行。BiMo检查有2种方式,分别为监督和有因检查。监督检查为日常检查,其对象通常是已完成的临床试验,检查内容主要是临床试验数据,检查临床试验机构执行FDA法规的情况。当FDA收到对临床试验的投诉或在其他相似临床研究中发现问题时,FDA将启动有因检查。有因检查的对象主要是正在进行中的临床试验,检查结果分为3个等级:不采取措施(no action indicated)、自愿采取措施(voluntary action indicated)和官方采取措施(official action indicated, OAI)。如FDA发现被检查者严重违反法规,检查结果等级定为OAI,并会发出无标题信、警告信等,日后还可能再次进行检查以确认问题已得到整改。值得注意的是,FDA的临床试验检查对象不仅仅是临床试验研究人员,还包括临床试验的申请人、监察员、伦理委员会和合同研究组织等。
2.3 报告制度
为保障受试者安全,FDA十分重视临床试验的安全性报告,要求申请人关注药品的任何安全性信息,这些信息不仅包括来源于本临床试验所发现的药品安全性相关信息,还包括来自于国内、外的药品安全性相关信息,来自于任何临床或流行病学研究、动物研究、上市后报告、科学文献报告、未出版的科学论文以及申请人以前未向FDA报告的国外监管机构发布的药品安全性相关信息。对任何与药品使用相关的严重的、意外的不良反应以及由动物实验所发现的可能对人体健康有危害的结果,申请人应在自初次获得信息的15 d内向FDA作出书面报告。对任何意料之外的致命的不良反应,申请人应在自初次获得信息的7 d内通过电话或传真向FDA报告。申请人应积极调查所收到的任何药品安全性信息,并将相关的后续信息尽快报告给FDA。申请人还需每年向FDA提交报告(在INDA生效周年日期之前的60 d内提交),报告正在进行和已完成的临床试验情况以及下一年的研究计划。
3 药品审评的沟通机制
在药品审评过程中,如何加强审评机构与各类专家、企业代表之间的沟通和协调是事关审评效率及其质量的重要因素。会议是FDA采用的审评方式之一,FDA鼓励在资源许可范围内召开有助于药品评估和解决药品相关科学问题的会议,其基本原则是保证对药品临床研究过程中出现的任何科学或医疗问题都可公开、全面、自由地交流。申请人可向FDA申请召开INDA前会议、Ⅰ期临床试验结束会议、Ⅱ期临床试验结束会议和Ⅲ期临床试验结束会议(新药上市申请前会议)。此外,申请人也可通过电话、电话会议、信件和电子邮件等方式与FDA进行交流。
4 FDA的审评资源
4.1 审评人员
FDA的药品审评采用以内部评价为主体、同时吸纳外部审评力量作为有益补充的方式,药品审评主要由CDER或CBER负责组织,同时设立专家委员会参与技术审评。专家委员会包括相关领域的研究者、科学家、消费者、患者代表以及制药企业代表,他们给出独立的科学技术方面的建议,对CDER提出的咨询问题进行表决,其结果虽不具有法律效应,但CDER在大多数情况下都会予以接受。
博士学位是成为CDER审评人员的基本条件,科学家则需至少2年的博士后工作经验。CDER設有培训与交流办公室,每年为员工安排各种培训,并邀请其内、外部的专家开设专业讲座,同时对培训效果进行定期评估。2003年起,CDER开始使用在线学习系统“知识中心”,后者不仅为员工提供学习资源,且也提供技能评估方法,同时可根据绩效需求诊断制定出适当的学习干预措施,包括网上课件、发展评估和非传统的学习机会等。
4.2 经费保障
FDA的药品审评曾因效率低下、耗时过长而饱受制药企业和民众的诟病。1992年,经美国国会授权,FDA颁布了《处方药收费法》(Prescription Drug User Fee Act, PDUFA),此法案的实施在促进药品审评工作方面起到了重要作用。PDUFA只有5年的执行期,届时如不再提案讨论,则其将自动废止。自1992年至今,PDUFA已被修定至第Ⅴ版。
根据2005年FDA发表的白皮书,经过2个5年的实施,FDA的药品审评经费状况得到了较好的改善,使得在自实施PDUFA以来FDA批准的1 000多种新药和近100种新生物制剂中,50%的新药由美国最先予以批准,而此比例在实施PDUFA之前仅为8%。2018财年,FDA的财务预算总额为54亿美元,其中约24亿美元来自于各类申请收费。
自药品审评和注册收费以来,FDA的工作也有了以下几项重要改进:①增加了审评人员和工作人员,药品审评速度显著加快;②改进了信息管理、工作程序及其标准,使得药品审评工作更为严格、统一规范并具可预见性;③通过制定技术指导原则,帮助企业最大限度地减少不必要的研究项目,减少不必要的重复工作。这些改进提高了药品研发效率,降低了药品研发费用,缩短了药品审评时间。
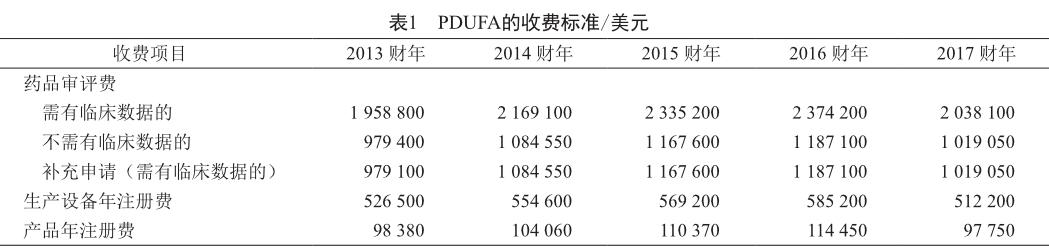
PDUFA授权FDA的收费项目有3种,分别为药品审评费(application fee)、生产设备年注册费(establishment fee)和产品年注册费(product fee)。对列为药品的生产设备,FDA按年度收取生产设备检查费用,即生产设备年注册费;对列入FDA产品目录的产品,FDA收取产品年注册费。药品审评费主要包括新药上市申请、生物制剂上市申请和补充申请费用,但不包括仿制药和血液制品等。不过,自2005年后FDA的仿制药审评效率受到业界质疑,待审量逐年积压,实际审评时间逐年延长。为此,2012年10月,美国国会又通过了《仿制药收费法》(Generic Drug User Fee Act),规定制药企业须向FDA支付仿制药上市申请审评费和生产设备检查费。
PDUFA的收費标准不是一成不变的,FDA在每个财年开始时都会根据通货膨胀率和审评工作量的不同对收费标准进行调整。自1993年PDUFA实施以来,PDUFA的收费标准逐年提高,收费总额也逐年增加,但2017财年的收费标准大幅降低(表1)。
5 对我国临床试验申请审批的借鉴
5.1 强化以安全性评价为中心的审批理念
我国的药品研发长期以来以仿制药研发为主,临床试验申请审批的理念多少还带着仿制药审批的影子,在考虑药品安全性的同时对基础药学研究看得过重,在对申请资料的要求上也重药学而轻临床。近年来,以安全性评价为中心的审批理念已逐步为我国所接受。在此基础上,我们应该逐步调整和完善相应措施,增加申请资料提交的灵活性,如简化药学部分申报材料、强化对临床资料的要求等。
5.2 加强对临床试验过程的监管
我们可以借鉴FDA的报告制度,要求申请人提交安全性报告,一方面促进申请人关注受试药品在临床试验中的安全性问题以及跟踪国内、外的最新安全性信息,另一方面有助于药监部门及时获得相关信息,通过风险分析及时发现潜在风险,保障受试者的安全。此外,也应要求申请人提交年度报告,以了解临床试验的进展情况,及早发现潜在问题并及早予以解决。
国内部分药品研发项目仅以申请得到临床批件为终点,并不真正进行临床试验。此类申请既占用了审批资源,又无实际科学意义。FDA要求申请人在INDA资料的封面上明确承诺“INDA生效后即开始临床试验”。我们可以借鉴此要求,要求申请人承诺进行临床试验,并设定相应的罚则,如3年内未进行临床试验的申请人不得再申请临床批件等。
5.3 建立有效的沟通机制
申请人与审批机构之间的有效沟通是提高审批效率的重要手段。FDA每年都会花费大量的时间用于这类沟通,但FDA认为此有助于药品审评。我国药品审批机构通过组织业务技术培训、设定开放日和咨询日,增加了药品审批相关信息的沟通渠道。但从申请人需求的角度来讲,这些沟通渠道仍显不足,我们应建立与申请人之间的多种形式的沟通、交流和对话途径,保证审批过程公开、透明。
参考文献
[1] 杨志敏, 杜晓曦. 中、美药品注册管理法规体系的比较研究[J]. 食品与药品, 2009, 11(1): 1-4.
[2] Levinson DR. The Food and Drug Administrations oversight of clinical trials [EB/OL]. [2018-05-03]. https://oig.hhs.gov/ oei/reports/oei-01-06-00160.pdf.
