反向胶束电动毛细管色谱法同时测定辣椒粉和豆制品中酸性橙Ⅱ和碱性橙2
2019-02-20,3
,3
(1.北京市疾病预防控制中心,北京 100013;2.北京市预防医学研究中心,北京 100013;3.首都医科大学公共卫生学院,北京 100069)
1 引言
非食用色素的滥用已成为食品安全的主要隐患。我国原卫生部自2008年以来陆续发布了5批食品中可能添加的非食用物质和易滥用的食品添加剂名单[1],碱性橙2和酸性橙II因属偶氮类碱性工业染料,均为中等毒性致癌化合物,已明确禁止在食品中使用[2],但由于二者着色牢固且不易被水浸泡出,价廉,高温下稳定等特点,较少的添加量即可达到理想的着色效果,一些不法商贩将其非法添加到辣椒粉、卤制品及豆制品中,使其在卖相上更加吸引消费者,而且二者联合添加,效果会更好,在辣椒粉中有被检出的报道[3],迫切需要建立这两种物质同时测定的分析方法,以确保食品安全。
目前针对两种物质的检测方法最常用的有高效液相色谱法(HPLC)[4-7]、LC-MS[3, 8-16]、GC-MS[17,18]等。这些方法灵敏度高、选择性好并能够对化合物进行确认,但其冗长的样品前处理:或固相萃取法,或浓缩富集,或低温下冷冻2h,都将导致检测时间的延长。现有的国标法[4]仅能检测碱性橙2,且其加标回收率低。
特别适合极性小分子物质分析的毛细管电泳法(CE)具有高效、快速、简便、耗样量小、溶剂消耗少、柱效高等特点,已用于工业染料的分析[20-24],但未见CE同时分离碱性橙2和酸性橙II的文献报道。本研究的目的在于建立一种同时检测辣椒粉和豆制品中非法添加的酸性橙Ⅱ和碱性橙2的反向MEKC方法,为食品卫生安全提供技术支持。
2 实验部分
2.1 仪器和试剂
Beckman PA 800 plus 型毛细管电泳仪,配PDA检测器;Beckman P/ACE Station 工作站(贝克曼库尔特有限公司,美国);未涂层熔融石英毛细管(河北邯郸鑫诺光纤色谱有限责任公司);Vortex-Genie 2涡旋混合器(美国Scientific Industries);11B S25 型研磨机(IKA,德国);Milli-Elix/RiOs超纯水仪(美国Millipore公司);F-50A酸度计(北京屹源电子仪器科技公司);Mettler Toledo电子天平(梅特勒-托利多,瑞士);德国Hettich Universal 32离心机高速离心机。
十二烷基硫酸钠(SDS,纯度≥99%,Sigma-Aldrich,USA);十六烷基三甲基溴化铵(CTAB,99%, Sigma-Aldrich,USA);乙腈(色谱纯, DIMA公司);H3PO4(分析纯,国药集团化学试剂有限公司);NaH2PO4(分析纯,国药集团化学试剂有限公司);NaOH(优级纯,北京化学试剂公司);冰乙酸(CH3COOH,纯度≥99.8%,国药集团化学试剂有限公司);实验室用水为超纯水。
标准品:酸性橙Ⅱ(Acid OrangeⅡ,纯度≥98%,Sigma-Aldrich公司)。碱性橙2(Basic Orange2,纯度≥98.2%,北京振翔工贸有限公司)。
实验中的样品购于北京郊区农贸市场、当地超市和早市。阳性腐竹样品为本实验室自制。
2.2 标准储备液的配制
称取折算纯度后的酸性橙Ⅱ和碱性橙2标准品,分别放入10 mL容量瓶中,加入适量超纯水溶解、稀释后,再加入超纯水至刻度,充分混匀,配制1.0 g/L标准储备液,置4℃冰箱冷藏保存。
2.3 电泳条件
未涂层石英毛细管:75 μm (70 cm(有效长度:60 cm),样品温度:25°C;分离缓冲溶液:30 mmol/L NaH2PO4+ 30 mmol/L H3PO4+ 30 mmol/L SDS + 0.5 mmol/L CTAB;分离电压:-17 kV;进样压力及进样时间:0.5 psi,12 s;检测波长:酸性橙Ⅱ和碱性橙2分别为490 nm和460 nm。分离缓冲溶液:30 mmol/L NaH2PO4+ 30 mmol/L H3PO4+ 30 mmol/L SDS + 0.5 mmol/L CTAB;样品提取溶液:以60%乙腈与20%乙酸为样品提取溶液,但进样时溶液需用超纯水稀释,即最后含有15%乙腈与5%乙酸。
为保证校正峰面积和迁移时间的重现性,新装毛细管使用前在20 psi压力下,分别用1 mol/L NaOH溶液冲洗20 min;超纯水和分离缓冲液均冲洗5 min。每次进样前依次用1 mol/L NaOH溶液、超纯水和分离缓冲液分别冲洗3 min、3 min、2 min,从而保证结果的重现性。
2.4 样品处理
2.4.1 辣椒粉样品
称取辣椒样品大约0.5 g于15 mL离心管中,分别加入1 mL冰乙酸、3 mL乙腈,加入超纯水至5 mL刻度,涡旋混匀2 min,离心5 min,上清液需用超纯水稀释4倍,方可进样。
2.4.2 腐竹样品
腐竹样品为本实验室自制,具体方法:将腐竹样品用研磨机粉碎、加入适量碱性橙2标准溶液,充分混匀,冷冻干燥后备用。需要时称取腐竹样品约0.5 g于1.5 mL离心管中,依次加入200 μL冰乙酸、600 μL乙腈和超纯水共计1 mL,涡旋混匀后,离心5 min,上清液需用超纯水稀释4倍,方可进样。
2.4.3 豆制品(豆泡、豆腐干、豆腐皮)
将豆制品用研磨机打碎,装入50 mL离心管中,放入冰箱(-18℃)冷冻备用。需要时准确称取豆制品约0.5 g于1.5 mL离心管中,加入200 μL冰乙酸、600 μL乙腈和超纯水共计1 mL,涡旋混匀后,离心10 min,上清液需用超纯水稀释4倍,方可进样。
3 结果与讨论
3.1 检测波长的选择
文献报道的酸性橙Ⅱ和碱性橙2的检测波长一般分别在450 nm~490 nm和430 nm~490 nm。本研究将酸性橙Ⅱ和碱性橙2的标准储备溶液分别用样品提取液稀释成100 mg/L,在190~600 nm内进行紫外-可见吸收光谱扫描,酸性橙Ⅱ和碱性橙2分别在490 nm和460 nm处有明显吸收,如图1a和图1b所示。利用二极管阵列检测器可实现两种组分在其最大吸收波长处被检测。
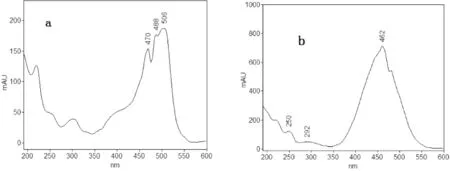
图1 在线扫描的酸性橙Ⅱ和碱性橙2的紫外-可见吸收光谱图
3.2 分离缓冲体系的选择
有研究选择pH 2.5的磷酸和磷酸二氢钾为分离缓冲体系[24],但在本研究初期,采用磷酸和磷酸二氢钠作为分离缓冲溶液时,20 min内并未见待测物峰。其原因为:酸性橙Ⅱ属偶氮类磺酸染料带负电,碱性橙2为偶氮类染料,其pKa=3.1,在pH 2.5时被质子化而带正电。故带负电的酸性橙Ⅱ因与电渗流(EOF)的方向相反而无法迁移到负极端被检测,而带正电的碱性橙2因pH 2.5时EOF低,导致其迁移到检测窗口的时间比较长,在可接受的20 min未能出峰。
受本实验室以前研究[25]的启发,往磷酸和磷酸二氢钠缓冲液中同时加入SDS和CTAB后,电渗流(EOF)反向,结果在短时间内检测到待测物的峰;可能的原因是:加入阴离子表面活性剂SDS后,形成胶束,将带负电的酸性橙II和带正电的碱性橙2包裹在胶束中,使待测物表面带负电荷,当加入EOF改性剂CTAB后,EOF反向,使待测物与EOF的方向一致,迁移到检测窗口被检测。0.5 mmol/L CTAB足以使EOF反向,且稳定性好[26],故选择0.5 mmol/L CTAB。故本研究选用H3PO4-NaH2PO4体系作为分离缓冲溶液。
3.3 分离缓冲溶液浓度的选择
本研究虽然只分析酸性橙Ⅱ和碱性橙2,但针对的样品一般为基质复杂的辣椒粉和豆制品,故本研究选用同时含有酸性橙Ⅱ和碱性橙2的辣椒粉样品进行分离条件的优化。一般情况下,分离缓冲溶液中缓冲盐的浓度高低影响着EOF的大小,盐浓度越高,EOF越低,迁移时间就会延长,分离度增加,而盐浓度过高会产生大量的焦耳热,使峰形变宽。分别考察了20、30和40 mmol/L NaH2PO4溶液对酸性橙Ⅱ和碱性橙2的迁移时间和分离度的影响,如图2所示,随着NaH2PO4浓度的增高,迁移时间不断延长,但分离效率增加,碱性橙2的灵敏度在40 mmol/L NaH2PO4时最高,但酸性橙Ⅱ的反而降低。综合考虑两者的分离度和灵敏度,30 mmol/L NaH2PO4为最佳选择。
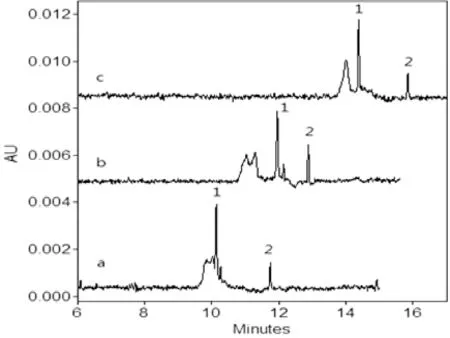
图2 分离缓冲溶液中NaH2PO4浓度对辣椒粉样品中两种待测物质分离的影响NaH2PO4浓度(mmol/L)a. 20;b. 30;c. 40;其它电泳条件见2.3;出峰顺序:1.碱性橙2;2. 酸性橙Ⅱ
3.4 分离缓冲溶液pH的优化
分离缓冲溶液的pH在磷酸盐的缓冲范围(pH 2.12±1)时,缓冲体系有较好的缓冲容量,才能保证良好的定性及定量重现性。本研究中,保持30 mmol/L NaH2PO4、30 mmol/L SDS和0.5 mmol/L CTAB不变的前提下,分别考察了pH值为2.3、2.25和2.2对分离的影响,相对应的H3PO4浓度分别为20、30和40 mmol/L。随着H3PO4浓度的增加,迁移时间逐渐延长,两者分离度和灵敏度得到明显改善,但是,碱性橙2与基质的分离并未随着H3PO4浓度的增加而显著增加,但分离时间显著延长,如图3所示。故在保证分离度的前提下,最终确定pH为2.25,相应磷酸浓度为30 mmol/L。
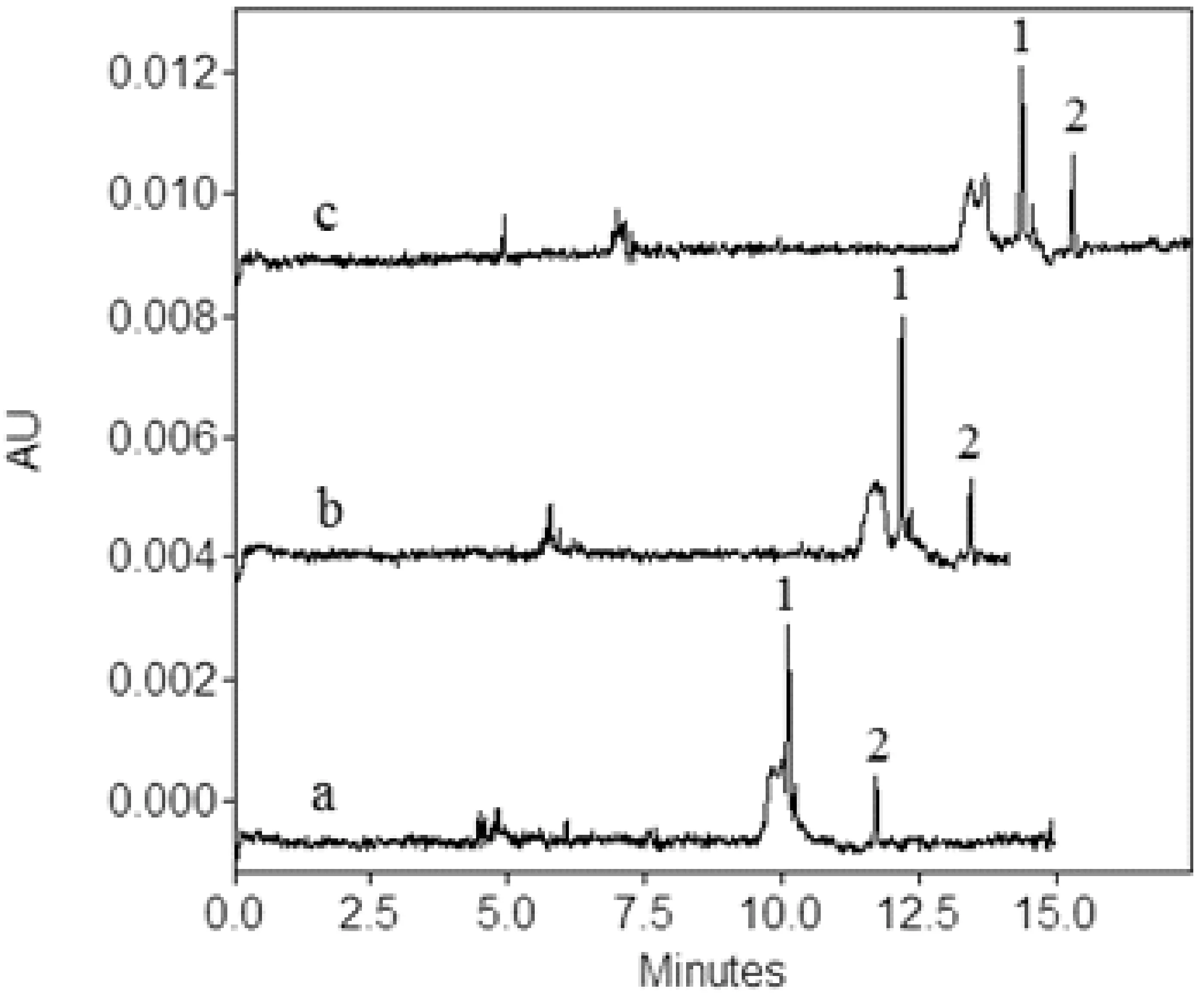
图3 分离缓冲溶液pH对辣椒粉样品中两种待测物质分离的影响pH a. 2.3;b. 2.25;c. 2.2;其它电泳条件见2.3;出峰顺序:1.碱性橙2;2. 酸性橙Ⅱ
3.5 分离缓冲溶液中十二烷基硫酸钠浓度的优化
分离缓冲溶液中适当加入阴离子表面活性剂SDS,不仅可以改善分离,还有增溶增敏的作用[27]。保持30 mmol/L NaH2PO4、30 mmol/L H3PO4和0.5 mmol/L CTAB不变,对SDS浓度分别为20、30和40 mmol/L进行了研究。结果表明,SDS浓度分别为20和40 mmol/L时,碱性橙2与样品基质不能分离,且酸性橙Ⅱ的灵敏度明显低于30 mmol/L SDS,基于分离度和灵敏度的考虑,最终选择30 mmol/L SDS为最佳,如图4所示。
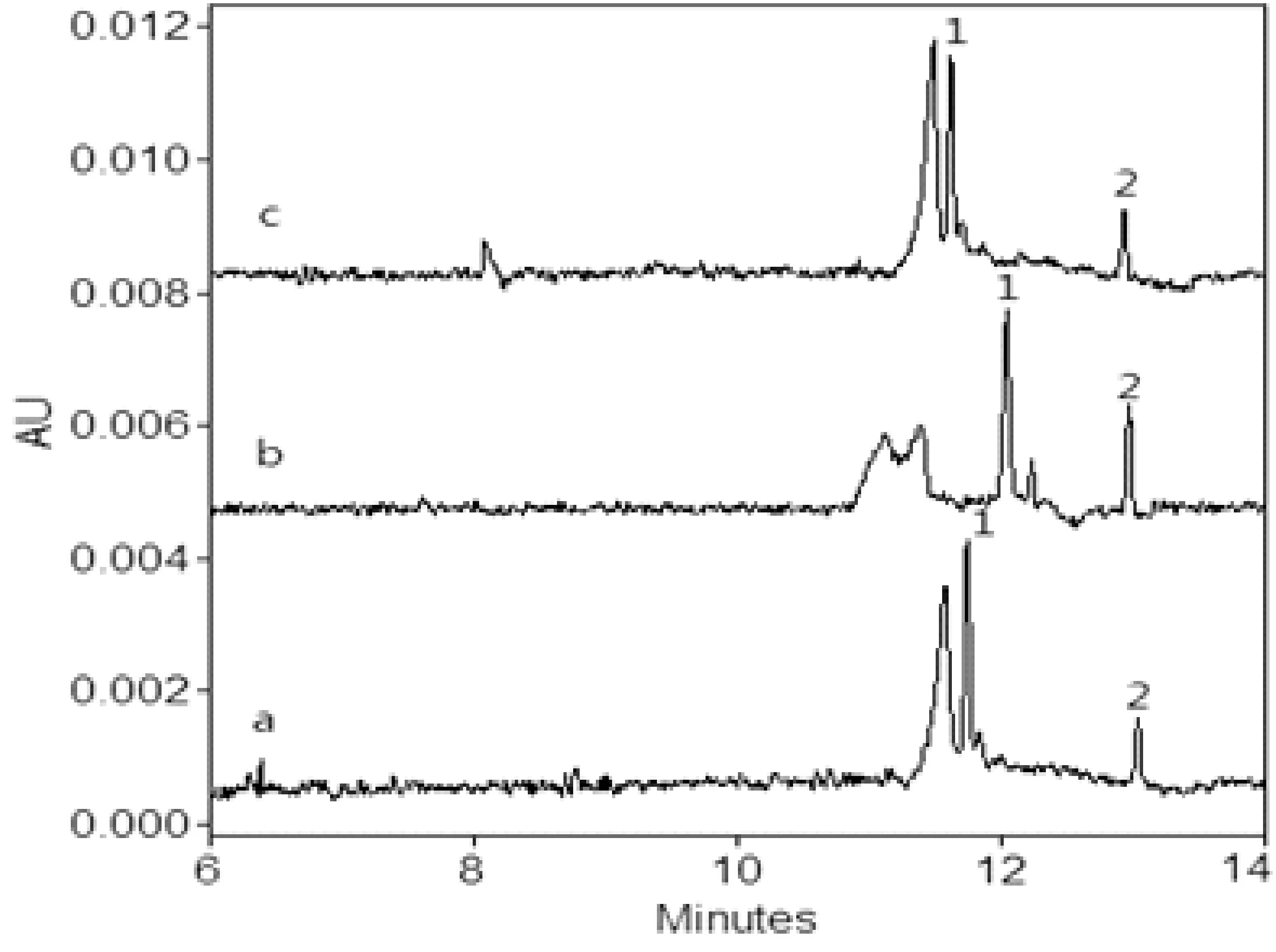
图4 分离缓冲溶液中SDS浓度对辣椒粉样品中碱性橙2和酸性橙Ⅱ分离的影响SDS浓度(mmol/L)a. 20;b. 30;c. 40;其它电泳条件见2.4;出峰顺序:1. 碱性橙2;2. 酸性橙Ⅱ
3.6 样品提取溶液的选择
染料的着色能力强,导致样品前处理方法在染料的准确测定中起着非常关键的作用。根据文献[7]中的样品前处理方法,选用水-乙腈-乙酸为提取液,在本试验过程中发现,随着乙腈和乙酸含量的同比例增高,乙腈层的颜色越深,提取得更彻底,经试验,选择含60%乙腈和20%乙酸为提取液可有效提取碱性橙2,然而含有如此高含量有机溶剂的样品溶液直接进样,易造成仪器报错,断电等情况而导致仪器运行停止,将此提取液用超纯水稀释4倍后进样,即进样时样品提取液含15%乙腈和5%乙酸,既能取得较高灵敏度和分离度,也能保证实验过程中不断电而使实验顺利进行。
3.7 分离电压的选择
分离电压直接影响着分析时间的长短,设置的电压越高,待测物迁移就越快,分离时间越短;但电压过高将产生大量焦耳热而导致峰展宽,灵敏度下降;而分离电压将使迁移时间增加,降低分析效率。经优化,-17 kV为最佳分离电压。
3.8 标准曲线、线性范围、精密度及加标回收率
3.8.1 标准曲线、线性范围、检出限及定量限
将1 g/L碱性橙2、酸性橙Ⅱ标准储备液用15%乙腈-5%冰乙酸依次稀释成质量浓度分别为1、2、4、8、16、32 mg/L以及2、4、8、16、32、64 mg/L的标准曲线,在上述MEKC条件下,由低到高质量浓度依次进样检测,用校正峰面积(峰面积除以迁移时间)外标法定量,以电泳峰的校正峰面积(A)为纵坐标,与其对应的质量浓度(ρ, mg/L)为横坐标,绘制标准曲线。线性范围、r、检出限(LOD)及定量限(LOQ)见表1。
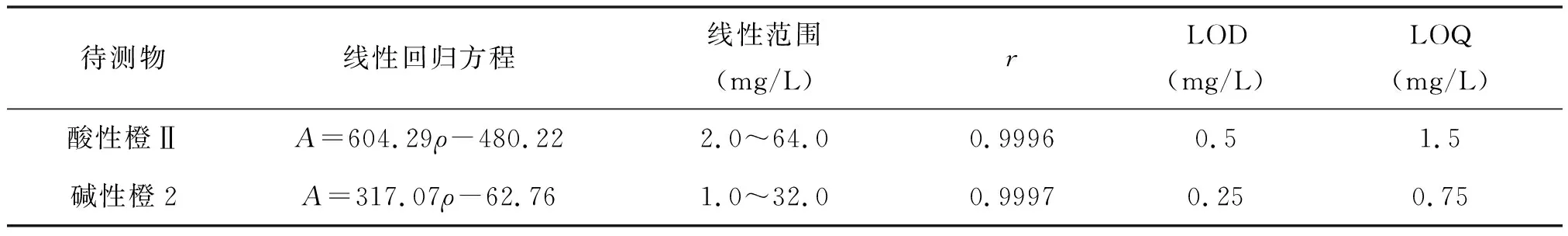
表1 线性范围、相关系数、检出限和定量限
3.8.2 仪器精密度
碱性橙2和酸性橙Ⅱ的仪器精密度测定分别在2、4、8 mg/L和4、8、16 mg/L3个质量浓度水平进行,每个质量浓度平行进样7次,计算迁移时间及校正峰面积的RSD%,结果见表2。
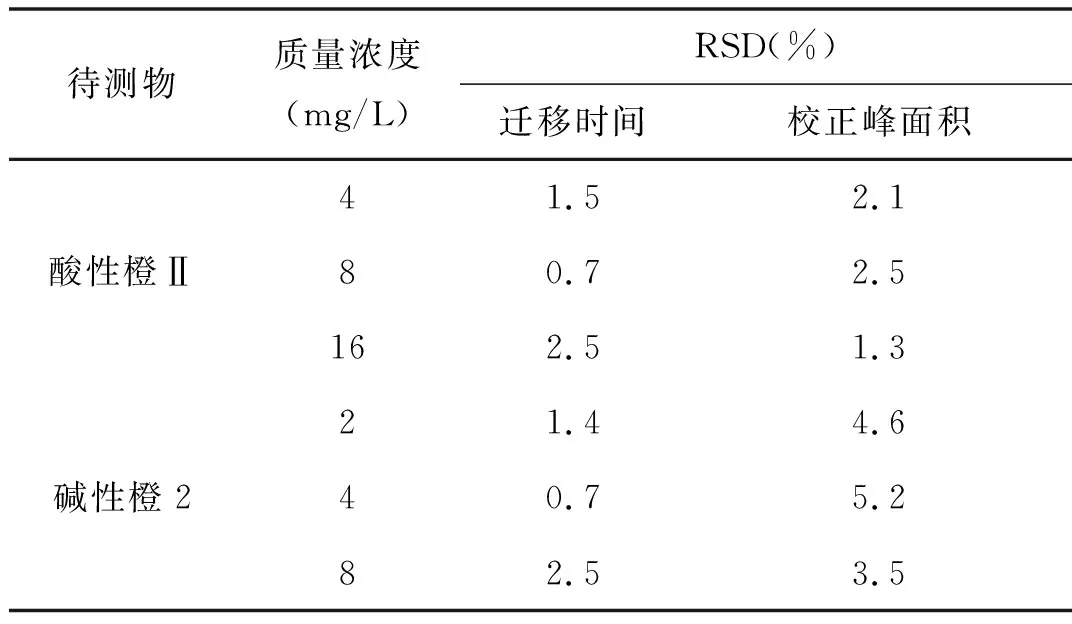
表2 仪器精密度(n=7)
3.8.3 方法精密度
方法日内精密度:按2.4中的处理方法,将辣椒粉样品,在一日内平行处理7份,在最优MEKC条件下进行测定,计算含量的RSD%,碱性橙2和酸性橙II的结果分别为4.7%和3.2%。
方法日间精密度:按2.4中的处理方法,在7个连续的工作日内,每日平行处理3份辣椒粉样品,得到当天质量浓度平均值,最后计算7天含量平均值的RSD%,结果分别为4.0%和4.5%。
3.8.4 加标回收率
选用阴性辣椒粉样品作为加标回收实验的本底,碱性橙2和酸性橙Ⅱ分别在2、4、8 mg/L以及4、8、16 mg/L3个质量浓度水平下进行加标回收实验的测定,3个质量浓度的样品平行处理5份,加入标准后室温放置过夜,使加标溶液与样品基质充分混合,第二天再行提取。所得低质量浓度加标电泳图如图5所示,加标回收率及RSD%列于表3。与文献[7]相比,本方法的回收率高,说明本方法对样品的提取更彻底。而HPLC法受色谱柱的限制,不能用太强的酸或碱进行提取,故回收率较本法低。
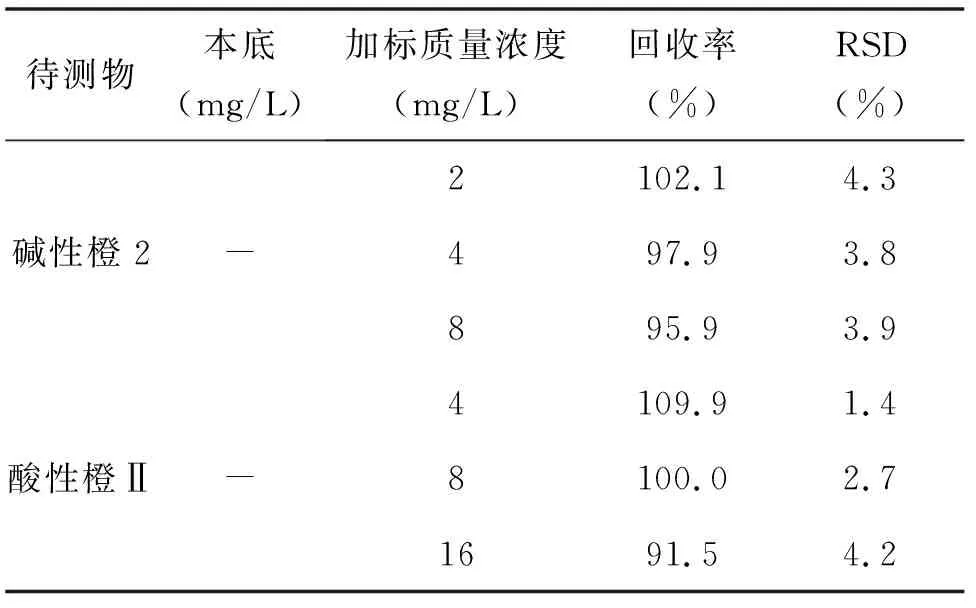
表3 加标回收结果(n=5)
-:未检出
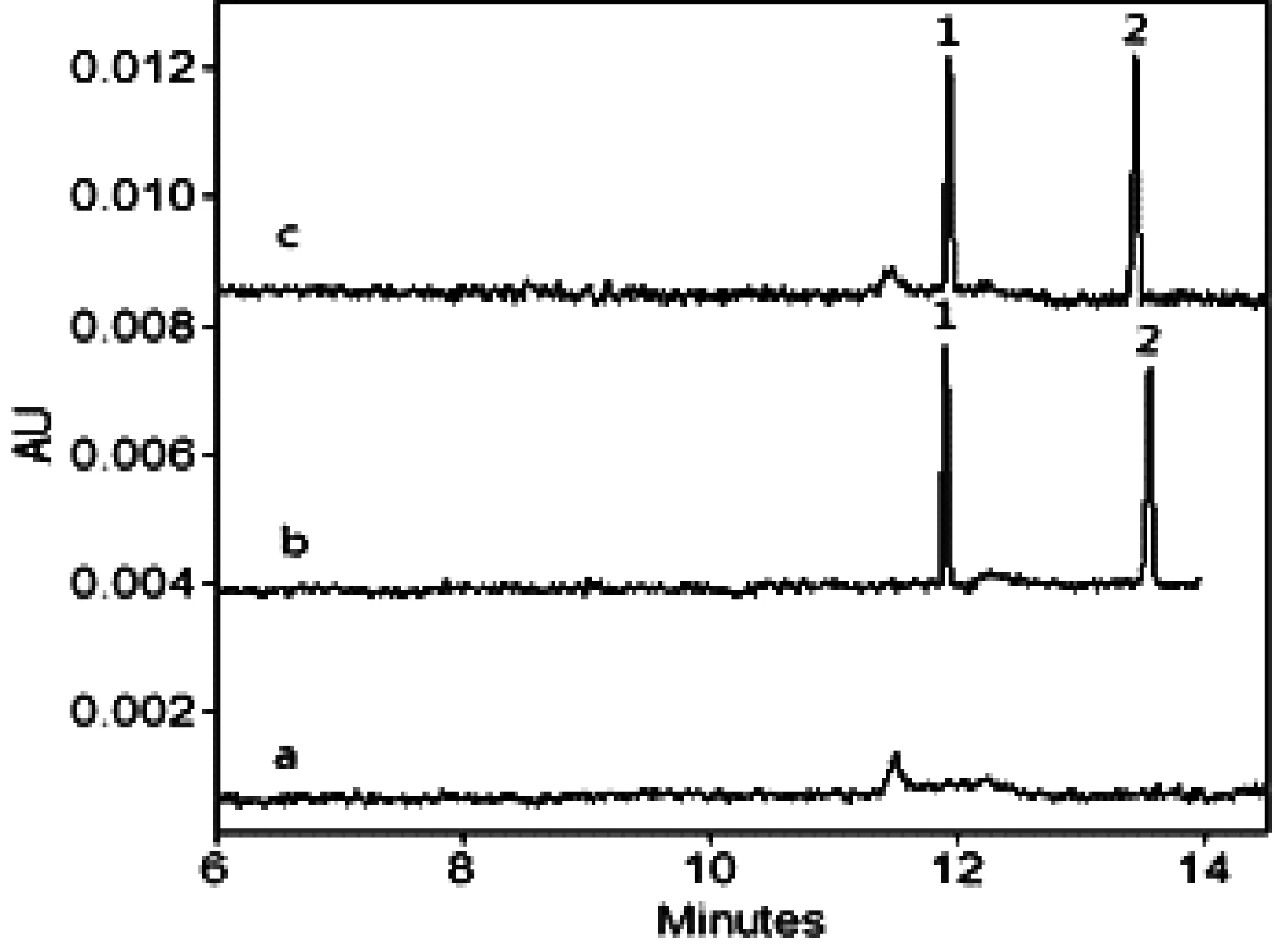
图5 辣椒粉样品低质量浓度加标回收电泳图a. 样品本底;b. 2 mg/L碱性橙2和4 mg/L酸性橙Ⅱ混标;c. 本底加标二混标;1. 碱性橙2;2. 酸性橙Ⅱ;其余电泳条件见2.3
3.8.5 样品分析
辣椒粉样品从北京农贸市场及超市购买,豆制品购买于早市,共计18件,分别编号后,依照文中2.4的方法进行样品前处理,每个平行处理5份进行测定,阳性样品含量及RSD%见表4,部分样品电泳图见图6、图7和图8。
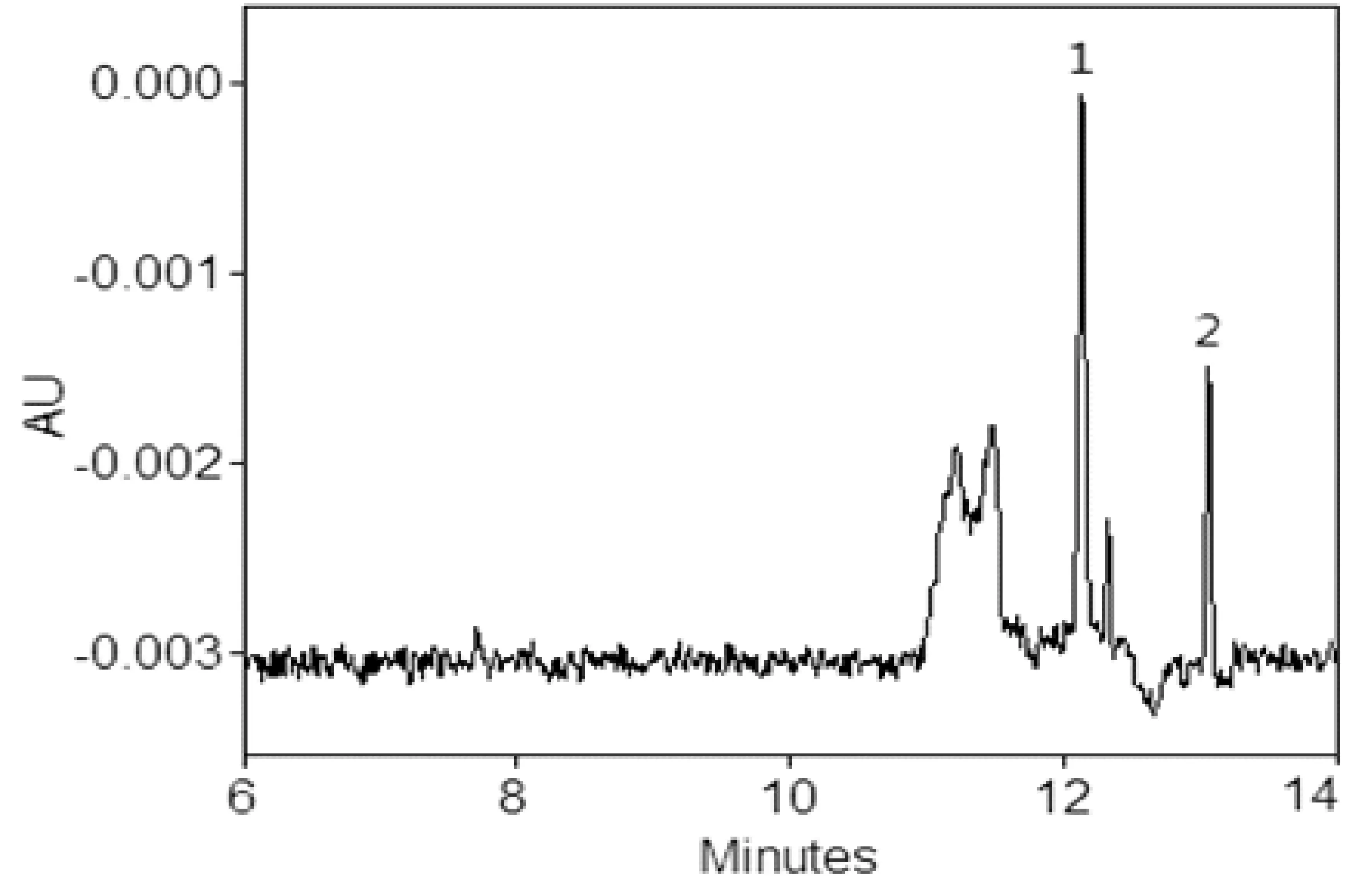
图6 1#辣椒粉样品电泳图1. 碱性橙2;2. 酸性橙Ⅱ;其它电泳条件见2.3
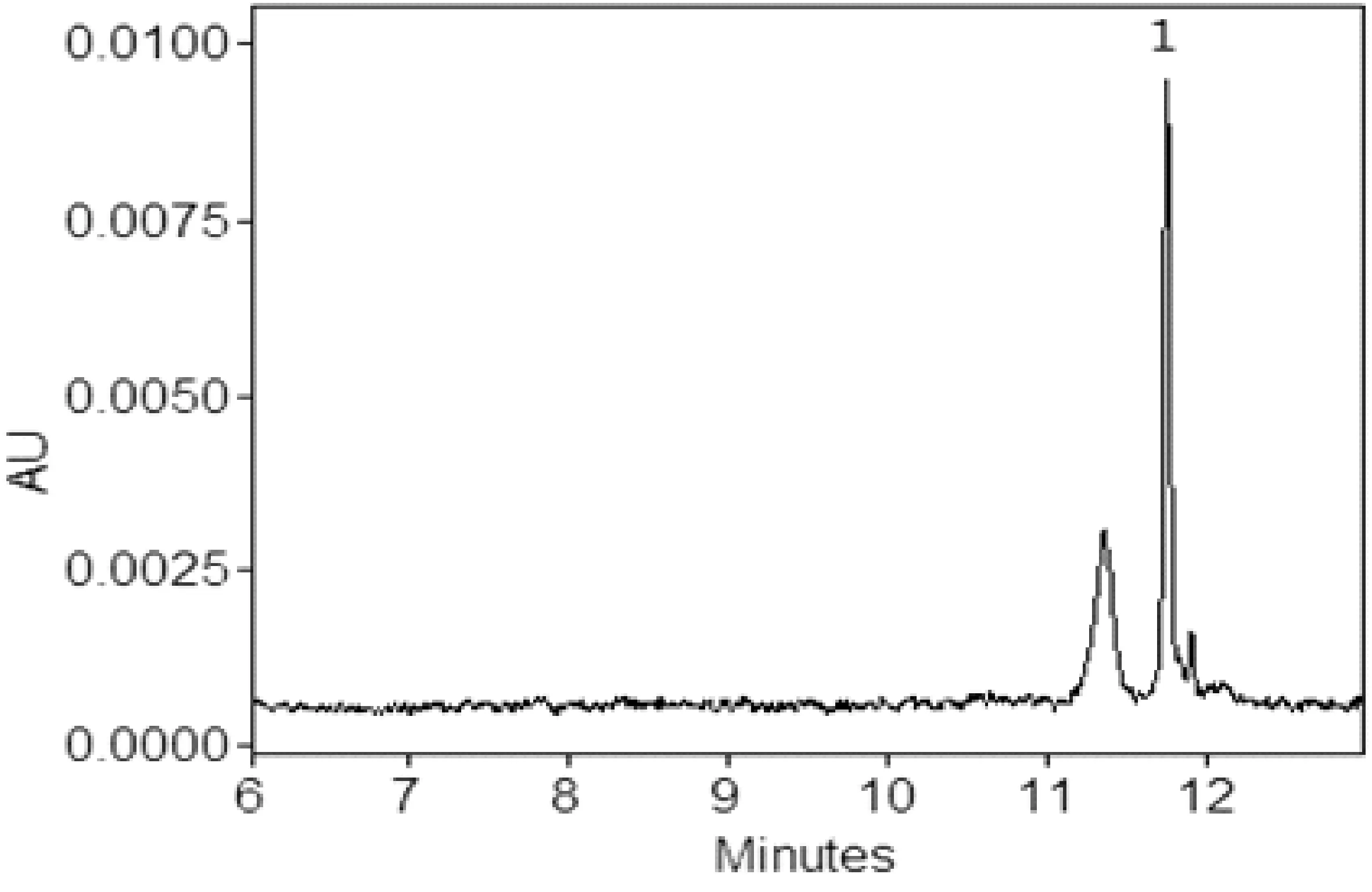
图7 2#辣椒粉样品电泳图1. 碱性橙2;其它电泳条件见2.3
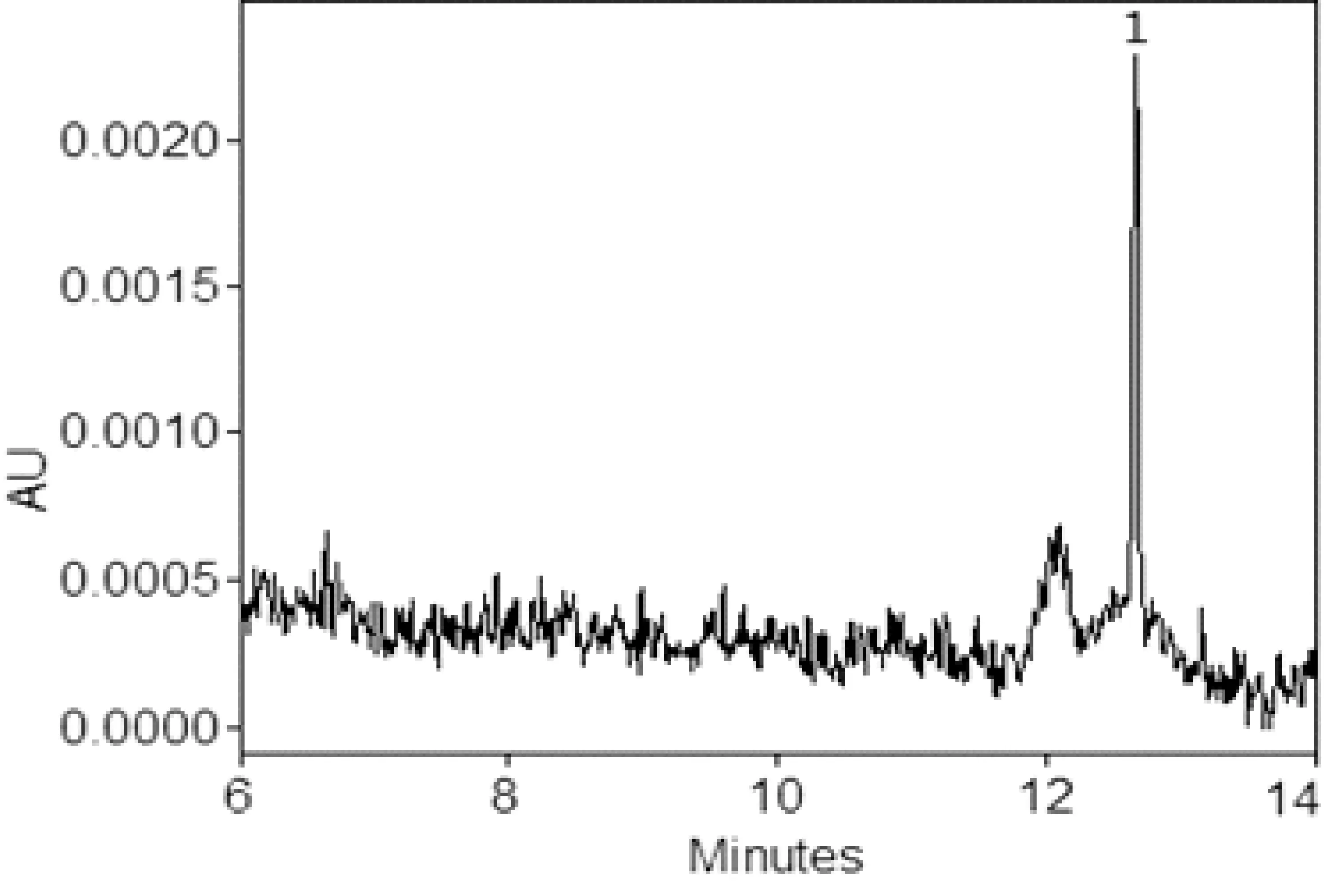
图8 9#腐竹样品电泳图1. 碱性橙2;其它电泳条件见2.3
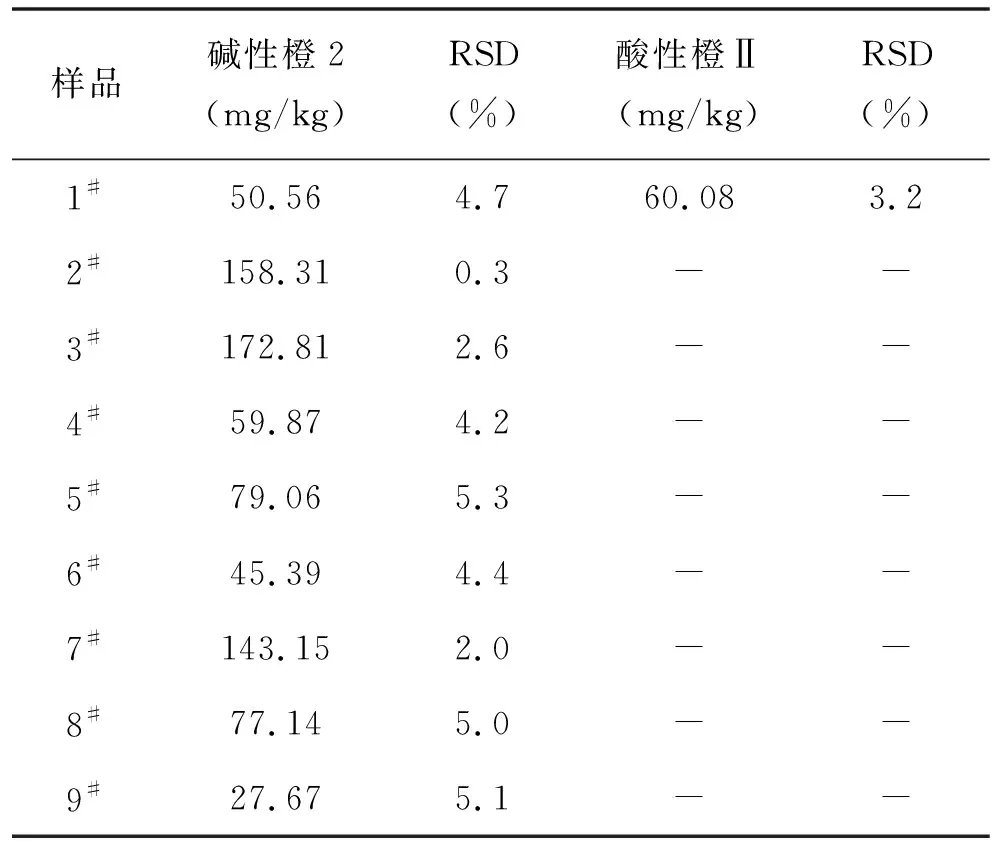
样品碱性橙2(mg/kg)RSD(%)酸性橙Ⅱ(mg/kg)RSD(%)1#50.564.760.083.22#158.310.3--3#172.812.6--4#59.874.2--5#79.065.3--6#45.394.4--7#143.152.0--8#77.145.0--9#27.675.1--
-:未检出
4 结论
建立了辣椒粉和豆制品中碱性橙2和酸性橙Ⅱ的反向MEKC分析新方法。选择含60%乙腈和20%乙酸水体系作为样品提取液有利于沉淀豆制品中蛋白;对于辣椒粉样品也有很好的提取作用,能够提高检测灵敏度,方法简单,省时省力,易于操作。加标回收率在91.5%~109.9%之间,解决了国标方法对碱性橙2回收率低的问题。方法的精密度均在6%以下,满足定性及定量分析要求。而HPLC法受色谱柱的限制,不能用太强的酸如本法中的提取液进行提取,导致HPLC法[7]的回收率较本法低,充分证明了本法优势,可避免因提取不完全而导致的假阴性结果。
