基于RAD-SNPs分析的四川核桃良种资源的遗传多样性研究
2019-02-13闫思宇朱鹏龚伟王景燕吴开志吴春艳李海平龚毅红段琼
闫思宇, 朱鹏, 龚伟*, 王景燕, 吴开志, 吴春艳, 李海平, 龚毅红, 段琼
基于RAD-SNPs分析的四川核桃良种资源的遗传多样性研究
闫思宇1*, 朱鹏1*, 龚伟1**, 王景燕1, 吴开志2, 吴春艳2, 李海平2, 龚毅红2, 段琼2
(1. 四川农业大学林学院, 成都 611130; 2. 四川省林木种苗站, 成都 610081)
为了解四川核桃()种质资源的遗传多样性,利用RAD二代测序技术对42份核桃良种资源进行测序,分析其遗传结构和遗传多样性。结果表明,共获得70G clean data,Q30平均为96.3%,并获得160 309个多态性SNPs。聚类、遗传结构和主成分分析结果高度一致,42份种质资源聚为两大类,基本反映了两个原始祖先血统,即普通核桃类(AJR,33份)和泡核桃类(AJS,9份),两类群具较高的遗传分化(ST=0.285),且分类与地理位置分布和种群相关;且修正了‘威薄01’、‘白龙1号’和‘石棉巨核’3个品种的类群认定。AJS和AJR类群中分别有9和14个良种为纯血统且亲缘关系较远,而其余的良种均为两个血统的混杂。42个核桃良种资源的核苷酸多样性[P()]和期望杂合度()分别为0.029和0.286,表明这些资源具有丰富的遗传多样性,相对于AJR类群,AJS类群的杂合率()与核苷酸多态性更大,而SNP数更少,AJS遗传多样性更丰富。这些为四川的核桃种质资源保存和杂交育种研究奠定了基础。
RAD-SNP;普通核桃;泡核桃;遗传多样性
植物种质资源是生产发展的基础性、战略性资源,是良种选育的原始材料、品种改良的物质基础,种质资源拥有的数量和研究的深入程度是决定育种效果的关键[1]。种质资源保护和保存的本质是遗传多样性的保护和保存,因此种质资源保护与保存是否有效,关键在于对种质资源的遗传类群、遗传关系、遗传多样性等遗传背景信息的研究和评估。核桃为胡桃科(Juglandaceae)核桃属()植物,具有用途多样、营养丰富、口味独特的特点,是世界著名的坚果类和木本油料树种之一[2–3],在人类健康和世界经济贸易发展中有着重要的作用[4]。核桃属含4组约21种,其中普通核桃()与泡核桃()为核桃组中的两姐妹种,是目前栽培最为广泛的两种核桃(作为干果食用)[5–7]。中国的核桃遗传资源十分丰富,各种类型和品种多达4 000多个,而经选育形成的优良品种或无性系就达到500余个[2]。四川是中国主要的核桃遗传资源分布和栽培省份之一,也是泡核桃和普通核桃共存的适生区域,蕴藏着丰富的核桃遗传资源[2,6]。近年来,随着一大批引进和本地选育优良核桃品种在生产中的使用,四川的核桃产业得到了快速的发展。为了更好地发挥核桃良种资源在生产和育种上的潜力,四川省林业厅组织全省相关的核桃研究和生产单位对目前四川范围内引进和选育的核桃良种资源进行了收集和保存,建立了四川省的核桃良种种质资源库,但要真正有效地利用和发挥这些核桃良种种质资源的遗传潜力,就需要对种质资源的遗传类群、遗传关系、遗传多样性等在内的遗传背景信息开展系统的研究和评估。
利用分子标记评价核桃遗传多样性,如限制性内切酶片段长度多态性标记[8](restriction fragment length polymorphism, RFLP)、随机扩增多态性DNA标记[9](random amplified polymorphic DNA, RAPD)、扩增片段长度多态性标记(amplified fragment length polymorphism, AFLP)[10]和微卫星DNA标记(micro- satellite DNA, SSR)[11],已得到广泛的使用。然而这些传统分子标记的位点数量普遍偏少,获得的遗传结构和遗传多样性信息有限[12]。单核苷酸多态性标记(single nucleotide polymorphisms, SNP)是二等位基因,代表基因组中最小的遗传变异单元,在基因组中密度高、分布均匀,数据统计简单、准确,不存在数据兼容性问题[13],被国际植物新品种保护联盟用于对农作物进行DUS (distinctness, uniformity and stability)测试。尤其是近年来测序技术的快速进步使得获取大量SNPs变得高效而低廉,限制性内切酶酶切位点DNA测序(restriction-site associated DNA sequencing, RAD-seq)技术就是其中之一,操作简单,性价比高,可获得数以万计的SNP标记,已广泛用于对非模式物种的研究[14–15]。为此,本研究采用RAD-seq技术对核桃良种种质资源进行测序,同时以NCBI上全长700 Mb的‘Chandler’为参考基因组序列(https://www.ncbi.nlm.nih.gov/genome/? term=Juglans%20regia)进行比对分析,通过获取多态性SNPs对四川省收集的42份核桃良种种质资源进行遗传背景信息分析和研究,期望验证种质资源库对核桃种类的认定,了解核桃种质资源的遗传构成、遗传关系和遗传多样性。通过确定品种准确的遗传分群,揭示品种间的遗传关系,为筛选并保存优良种质资源奠定基础,也为未来进一步的杂交育种减少盲目性,同时为四川地区核桃种质资源的遗传多样性及遗传背景的分析提供重要的参考价值, 对四川的核桃育种工作提供重要的理论指导。
1 材料和方法
1.1 材料
试验材料为经四川省林木品种审定委员会审(认)定的42个核桃良种(表1),其中6个为北方引进品种、3个为云南引进品种、4个为本地杂交品种,其余都是通过对四川野生或长期栽培的核桃进行选育的品种(http://www.sclmzm.com:88/sczmz/linmuliangzhong/index.jhtml)。北方品种是利用北方核桃直接选育或杂交后选育出的[16–17],云南品系为新疆核桃与云南的漾濞核桃(泡核桃)、三台核桃和华宁核桃等杂交选育出的云新系列[18–19],本地杂交核桃是云新系列与四川盆地本地核桃的杂交品种[20–22]。于2016年5月上旬在四川农业大学崇州基地分别采集各品种的嫩叶,采集前用100%酒精消毒、清洗,采集后放入冰盒保鲜,立即带回实验室进行DNA提取。
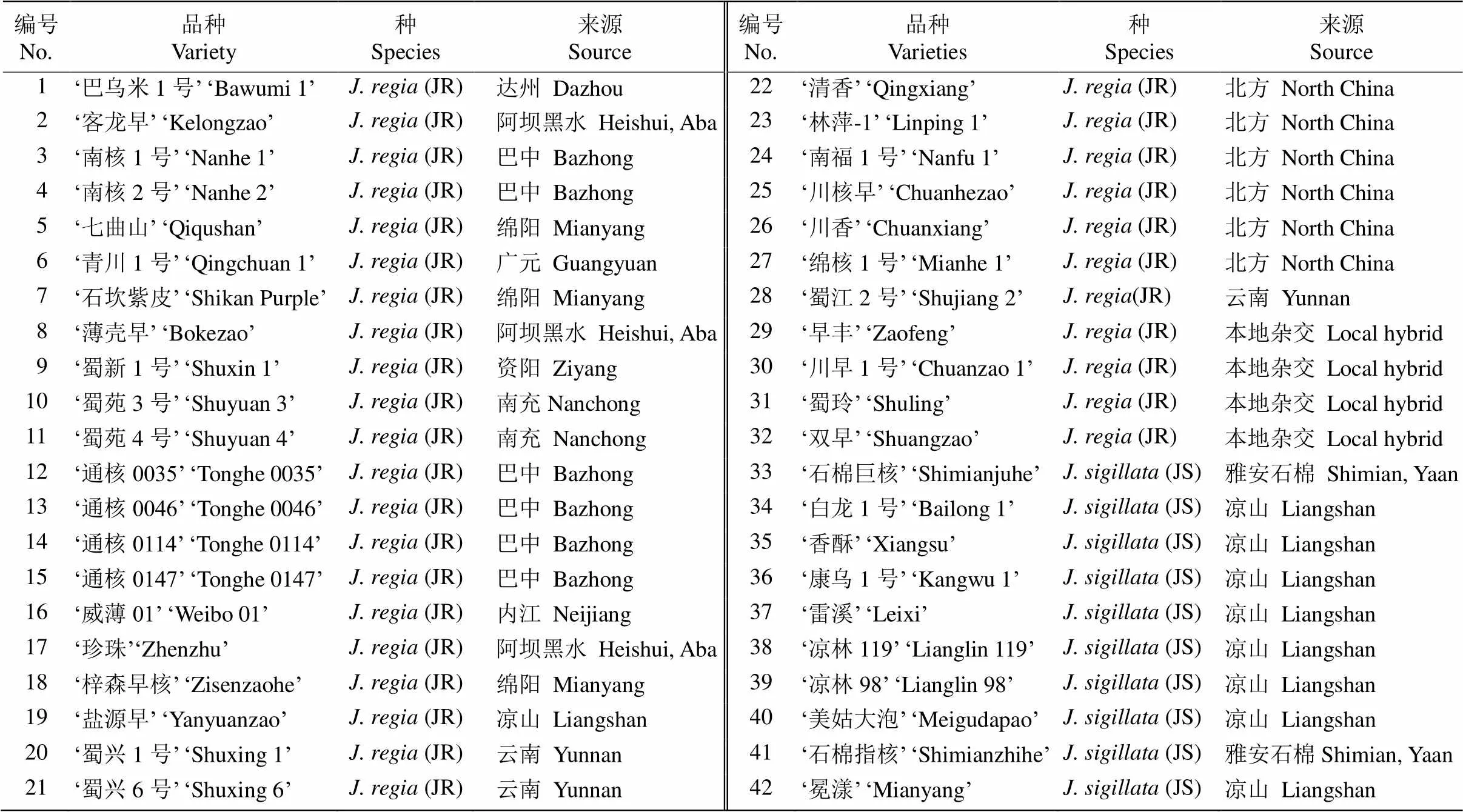
表1 42个供试核桃良种
1.2 基因组DNA的提取
加入4%~6%聚乙烯吡咯烷酮(polyvinyl pyrro- lidone, PVP)和液氮研磨嫩叶,采用宝生物植物DNA提取试剂盒(TaKaRa, Dalian)提取核桃基因组DNA。提取的DNA采用琼脂糖凝胶电泳和ND- 2000分光光度计检验DNA的质量和浓度,将合格的样品送于上海美吉生物医药科技有限公司进行RAD-seq简化基因组测序。
1.3 文库构建和测序
利用酶切预测软件对参考基因组进行酶切预测,选择酶切效果最佳的内切酶I(TCGA Thermo- fisher)进行后续实验。对检测合格的样品基因组DNA以总量800 ng参照Baird等[23]进行酶切文库构建,文库质检合格后用Illumina HiseqTM平台进行测序,测序策略为Illumina PE150。为评估建库实验的准确性,选用水稻()作为对照参与建库和测序。为了保证测序质量,对‘凉林119’与‘白龙1号’进行了两次测序,通过比对结果分析进行内部控制数据质量,因此共进行了44份材料的测序。测序数据进行质量过滤,并对过滤前后的测序reads数、总碱基数、Q30和平均测序深度进行统计。
1.4 SNP标记开发
运用BWA 软件[24]将质控后的reads与参考基因组序列进行比对,再利用GATK软件[25]消除假阳性SNP和Samtools软件[26]提供的vcfutils工具过滤掉测序深度和比对质量值较低的位点,过滤标准为:① 选取基因组中的非重复区(避免转座元件),并映射在参考基因组上的reads (避免非特异性扩增);② 只保留双等位基因频率范围在40%~60%的SNPs;③ 相邻的SNPs至少相隔50 kb;④ 筛选掉样品分型结果中次要等位基因频率MAF<0.05的位点;⑤ 筛选基因型至少覆盖所有样本60%以上个体的标记 (该标准根据实际标记数据量进行适当调整),即对于单个多态性标记位点,100个样本中至少有60个样本有确定基因型。通过对基因型完整性覆盖情况差的标记进行过滤,最终得到的SNP标记用于后续分析。
1.5 遗传结构分析
通过RAxML软件[27]的maximum likelihood算法构建进化树,使用模型为GTRGAMMA, bootstrap 1 000次;通过STRUCTURE v2.3.4[28]软件,分析遗传结构,假设样本的分群数(K值)为1~10,使用贝叶斯算法推算在具体分群数下每个个体的血统构成情况。根据CV error (cross validation error)最低点对应的K值来确定最佳分群数。通过GCTA软件[29]进行主成分分析(principal components analysis, PCA)分析,得到样本的主成分聚类情况,用于辅助遗传结构分析。
1.6 遗传多样性分析
根据对42个样本的遗传结构分析结果,对类群的遗传多样性进行分析,包括核苷酸多样性[P()]、群体期望杂合度()等。核苷酸多样性采用PopGenome软件[30]进行计算,期望杂合度为每个位点杂合样数目与所有样本数目之比,然后将所有位点的杂合率相加,取平均值。
2 结果和分析
2.1 测序结果和SNP开发
对44份材料进行测序,共获得70G clean data,Q30平均为96.3%,GC含量平均为43.0% (数据集1),样本测序深度为23×~64×(数据集3)。所有样本定位到参考基因组上的clean reads数占所有去重复clean reads数的88.6%,同时符合测序片段长度的reads数占所有clean reads的74.7% (数据集1)。
以核桃‘Chandler’品种为参考基因组序列进行比对,共获得7 360 659个SNPs (数据集2)。经过假阳性消除和基因型完整性覆盖过滤后,共获得160 309个高一致性的群体SNPs,其中有6 357个测序深度在所有样品中都超过10×。160 309个SNPs被定位到“Chandler”基因组序列(105811个)的4743个片段上,其中有1 116个SNPs定位到LIHL01055144片段,为含量最多的片段。基因间隔区、外显子、下游、上游和ncRNA外显子(ncRNA-exonic)分别含有的SNPs为158 049 (98.6%)、928 (0.6%)、479 (0.3%)、764 (0.5%)和89 (0.1%)。其中外显子含有的928个SNPs中,同义SNV (single-nucleotide variant)和非同义SNV分别为319 (34.4%)和375 (40.4%)(数据集4)。
2.2 遗传结构分析
聚类分析(图1)表明,42个品种和‘Chandler’品种比较明显地聚为两大类群:第一类群包含9个品种(标为AJS),除‘威薄01’外,其余品种均隶属于泡核桃[(JS)];‘Chandler’和另外33个品种聚为第二类群(标为AJR),除‘白龙1号’和‘石棉巨核’外,其余品种均隶属于普通核桃[(JR)]。基于Structure软件分析的遗传结构结果表明,当分类群数K=2时的CV-error最小(图2), 表明42个样品可能主要存在两类血统来源[18]。具体分析分类群数K=2时42个品种的血统来源(图3),AJS类群中的‘香酥’、‘康乌1号’、‘雷溪’、‘凉林119’和‘冕漾’等5个品种为完全的纯蓝色血统(标为AJS-2),其余4个品种混杂有部分紫色血统(标为AJS-1);AJR类群中有14个品种为完全的纯紫色血统(标为AJR-2),其余品种均为紫色和蓝色血统的混杂(标为AJR-1)。PCA分析结果与聚类分析、Structure分析结果具有相当的匹配性,42个品种也可分为两大类群4个亚群(图4),同时根据解释比例最高(占19.2%)的主成分PC1,4个亚群呈一定的梯度排列,依次为AJS-2、AJS-1、AJR-1和AJR-2,即表现出从纯正的祖先血统I→杂合血统→祖先血统II的梯度关系,AJS-2和AJR-2两亚群相距最远,遗传结构差异性最大,亲缘关系最远。
2.3 遗传多样性分析
遗传多样性分析表明,AJS类群的品种数虽然要少于AJR类群,但AJS类群的核苷酸多样性[P()]和期望杂合度(He)都高于AJR类群,AJS类群分别为0.030和0.308,AJR类群分别为0.027和0.278,而42份核桃种质的核苷酸多样性和期望杂合度介于两个类群之间,分别为0.029和0.286,具较高的遗传多样性。
3 讨论和结论
随着植物全基因组序列研究工作的广泛开展, SNP标记已开始广泛地应用于植物的遗传分析[31–32]。有研究表明,低等到中等RAD测序均可应用于非模式物种种群基因组的探索[4,23]。核桃是雌雄同株异花,多异交,可天然杂交,在种群内保持着多样化的基因型[2–3]。通过群体DNA池开发的SNP较从单个个体开发的标记对于研究遗传多样性的效果更佳[33]。本研究中,普通核桃与泡核桃通过共同使用RAD策略进行基因组测序,并对‘凉林119’和‘白龙1号’重复测序,从内外部进行数据质量控制,排除物种或限制酶干扰,从而保证测序质量。‘凉林119’和‘白龙1号’两次测序个体间的遗传距离存在边际效应(<0.005),这可能与分析处理中数据(不包括冗余数据)非对称丢失有关[14]。本研究中,每个样本测序数据量均较高(测序深度为23×~64×), 而160 309个高一致性的群体SNPs中有6 357个测序深度在所有样本中都超过了10×,高于Xu等[14]报道葫芦()的RAD-seq平均测序深度,因此对42个品种开展的遗传分析具有较好的可信度。
聚类分析结果表明,42个核桃品种和‘Chandler’品种聚为两大类群,结构分析也表明两个类群是最优的分类群数,且除内江威远的‘威薄1号’、雅安石棉的‘石棉巨核’和凉山会理的‘白龙1号’外,各品种也基本符合原有认定的普通核桃和泡核桃的分类。根据STRUCTURE和PCA的分析结果,两大类群又可分别分为两个亚群,即纯血统和混杂血统两个亚群,其中纯血统普通核桃为6个北方引进品种、2个阿坝黑水优选品种和其他一些盆地优选品种,纯血统泡核桃都是川西山地品种。可以看到,四川盆地的优选核桃除‘威薄1号’外均与此前的分类一致,更多的是属于北方核桃的普通核桃类群或血统。根据核桃的地理起源考证,普通核桃可能起源于西亚和我国北方地区(尤其是新疆地区),而后主要由于人为活动而被传播到现在的主要栽培区[3,34],四川盆地至少早在汉代以前就与北方地区有着十分频繁的交流,普通核桃也因此很可能作为优良的坚果经济林而被引入栽培,根据四川的地形地貌和生态气候条件,四川核桃传统上被分为东部盆地区的川东亚群和西部山地区的川西亚群[2],但本研究结果中阿坝黑水和广元青川核桃并没有和其他川西核桃聚为一个类群,而是与川东亚群和北方核桃等聚为一个类群。Wang等[35]在对我国8个野生核桃种群进行遗传分析时,也认为黑水的核桃种群与华北地区的核桃种群聚为一类,显示其与北方核桃有更近的血缘关系,这很可能也是因为历史上该地区与盆地地区比凉山地区有更为频繁的交流有关。雅安市南部大相岭以南的凉山地区是泡核桃的原产分布区之一[3,36],该地区优选的核桃除‘盐源早’、‘石棉巨核’和‘白龙1号’都归属于泡核桃类群,也基本符合此前的分类。‘盐源早’虽然优选于泡核桃原产分布区的凉山盐源县,但根据其形态特征以及盐源县近年的核桃栽培和发展历史,曾被认定为普通核桃[37],本研究予以证实。雅安石棉优选的‘石棉巨核’和凉山会理优选的‘白龙1号’此前虽然被认定为泡核桃,但它们的形态特征偏向于普通核桃而一直被怀疑[38],本研究无论是聚类还是血统分析结果表明,它们更应该属于普通核桃而非认定的泡核桃。在此前认定为普通核桃的品种中,内江威远优选的‘威薄1号’是唯一1个被归为泡核桃的品种,根据Structure的血统分析,其泡核桃血统超过了60%,因此该品系归于泡核桃比较合理[39]。
核苷酸多样性是反映物种种内DNA序列变异程度的重要参数[40–43]。模式植物拟南芥()基于334个SNP位点和水稻()基于111个SNP位点的核苷酸多样性指数分别为0.007和0.003 2[43–44],木本植物的核苷酸多样性指数多为0.001 86~0.016[45],挪威云杉()基于22个位点的核苷酸多样性指数为0.003 99[46],毛果杨()基于全基因组SNPs的核苷酸多样性指数为0.016[47],凹叶木兰()基于7个SNP位点的核苷酸多样性指数为0.017 8[41]。在本研究中,42个核桃品系基于简化基因组SNPs的核苷酸多样性指数达0.029,远高于许多木本植物,显示这42份核桃种质资源具有较为丰富的遗传多样性。在期望杂合度方面,本研究42个品种的总体期望杂合度()为0.278,与多种物种基于SNP的期望杂合度相比处于中上水平, 如短花针茅(, 0.131)、(0.269~0.303)[48]、葡萄(, 0.340)[49]等。对比Han[50]的研究结果,本研究中核桃群体期望杂合度相对更低,可能由于SNP的等位基因一般远少于SSR标记,因此基于SNP的期望杂合度也低于基于SSR标记的群体期望杂合度,与葡萄的研究结果相似[49]。泡核桃类群的样本数远低于普通核桃类群,但其核苷酸多样性(0.030)和期望杂合度(0.308)都要略高于普通核桃类群(0.027和0.278), 显示出更高的遗传多样性。相对于普通核桃,泡核桃的原产地区地理条件险峻恶劣,历史上经济发展也十分滞后,且泡核桃本身的食用品质较差,因此一般都是以野生方式存在,很少经历人为的驯化和选育,可能是导致泡核桃类群在个体数较少的情况下仍然具有较高遗传多样性的原因[51–52]。
本研究采用RAD-seq技术对42份核桃良种种质资源进行测序,获得160 309个高一致性的SNPs,遗传分析表明,42个核桃良种基本符合此前的分类认定,分为普通核桃和泡核桃两大类群,且两大类群又可细分为血统较纯和较混杂的两个亚群,其中普通核桃纯血统亚群主要包括北方引进品种和阿坝黑水品种等,泡核桃纯血统亚群都是凉山地区选育品种,而云南引进的杂交品种和本地杂交品种都属于普通核桃混杂血统亚群。根据遗传类群划分对3个核桃品种‘威薄01’、‘石棉巨核’和‘白龙1号’之前的类群认定进行了修正,‘威薄01’修正为泡核桃,而‘石棉巨核’和‘白龙1号’修正为普通核桃。从核苷酸多样性指数和期望杂合度来看,42份核桃种质资源具有较为丰富的遗传多样性,且泡核桃类群的遗传多样性略高于普通核桃类群。因此,该批良种种质资源可以为开展四川核桃育种提供良好的遗传资源和物质基础,同时也为四川省核桃种质资源的区划、保护及利用提供理论依据。
[1] ZHANG T. Research on genetic diversity of tobacco germplasm resource [J]. Serv Agric Technol, 2010, 27(1): 101-103. 张艇. 烟草种质资源的遗传多样性研究 [J]. 农技服务, 2010, 27(1): 101-103.
[2] XI R T, ZHANG Y P. Flora of China Fruit Tree, Volume Walnut [M]. Beijing: China Forestry Publishing House, 1996: 28-53,84-91.郗荣庭, 张毅萍. 中国果树志, 核桃卷 [M]. 北京: 中国林业出版社, 1996: 28-53,84-91.
[3] XI R T, ZHANG Y P. China Walnut [M]. Beijing: China Forestry Publishing House, 1992: 111-117. 郗荣庭, 张毅萍. 中国核桃 [M]. 北京: 中国林业出版社, 1992: 111–117.
[4] BAGHKHEIRATI E K, BAGHERIEH-NAJJAR M B. Modelling and optimization of Ag-nanoparticle biosynthesis mediated by walnut green husk extract using response surface methodology [J]. Mat Lett, 2016, 171: 166-170. doi: 10.1016/j.matlet.2016.01.159
[5] ARADHYA M K, POTTER D, GAO F Y, et al. Molecular phylogeny of(Juglandaceae): A biogeographic perspective [J]. Tree Genet Gen, 2007, 3(4): 363-378. doi: 10.1007/s11295-006-0078-5.
[6] PU G L, XIAO Q W, WU K Z, et al. Research on the phenotypic diversity of walnut germplasm resources in Sichuan [J]. J Hunan Agric Univ (Nat Sci), 2014, 40(2): 162-167. doi: 10.13331/j.cnki.jhau.2014. 02.011.蒲光兰, 肖千文, 吴开志, 等. 四川核桃种质资源表型多样性研究 [J]. 湖南农业大学学报(自然科学版), 2014, 40(2): 162-167. doi: 10. 13331/j.cnki.jhau.2014.02.011.
[7] PEI D, LU X Z. Walnut Germplasm Resources in China [M]. Beijing: Chinese Forestry Press, 2011: 25-28. 裴东, 鲁新政. 中国核桃种质资源 [M]. 北京: 中国林业出版社, 2011: 25-28.
[8] FJELLSTROM R G, PARFITT D E. Phylogenetic analysis and evolu- tion of the genus(Juglandaceae) as determined from nuclear genome RFLPs [J]. Plant Syst Evol, 1995, 197(1/2/3/4): 19-32. doi: 10. 1007/BF00984629.
[9] NICESE F P, HORMAZA J I, MCGRANAHAN G H. Molecular characterization and genetic relatedness among walnut (L.) genotypes based on RAPD markers [J]. Euphytica, 1998, 101(2): 199-206. doi: 10.1023/A:1018390120142.
[10] BAYAZIT S, KAZAN K, GULBITTI S, et al. AFLP analysis of genetic diversity in low chill requiring walnut (L.) genotypes from Hatay, Turkey [J]. Sci Hort, 2007, 111(4): 394-398. doi: 10.1016/ j.scienta.2006.11.006.
[11] DANGL G S, WOESTE K, ARADHYA M K, et al. Characterization of 14 microsatellite markers for genetic analysis and cultivar identification of walnut [J]. J Amer Soc Hort Sci, 2005, 130(3): 348–354.
[12] WANG W, ZHOU W Z. Efficient extraction and application of total RNA in sisal [J]. Chin J Trop Crops, 2008, 29(6): 725–729. doi: 10. 3969/j.issn.1000-2561.2008.06.010.王尉, 周文钊. 剑麻核酸的高效提取及应用[J]. 热带作物学报, 2008, 29(6): 725–729. doi: 10.3969/j.issn.1000-2561.2008.06.010.
[13] LI X, TIAN H L, WANG F G, et al. Comparison of SSR and SNP markers in maize varieties genuineness identification [J]. Mol Plant Breed, 2014, 12(5): 1000–1004. doi: 10.13271/j.mpb.012.001000.李雪, 田红丽, 王凤格, 等. SSR和SNP两种标记技术在玉米品种真实性鉴定中的比较分析 [J]. 分子植物育种, 2014, 12(5): 1000– 1004. doi: 10.13271/j.mpb.012.001000.
[14] XU P, XU S Z, WU X H, et al. Population genomic analyses from low- coverage RAD-Seq data: A case study on the non-model cucurbit bottle gourd [J]. Plant J, 2014, 77(3): 430–442. doi: 10.1111/tpj.12370.
[15] van WYNGAARDEN M, SNELGROVE P V R, DIBACCO C, et al. Identifying patterns of dispersal, connectivity and selection in the sea scallop,, using RAD-seq derived SNPs [J]. Evol Appl, 2017, 10(1): 102–117. doi: 10.1111/eva.12432.
[16] CHEN S B, YU Z J, YANG W Y, et al. Introduction and cultivation techniques of Qingxiang walnut in Sichuan [J]. S China Fruit, 2016, 45 (2): 156–160. doi: 10.13938/j.issn.1007-1431.20150532. 陈善波, 余忠江, 杨文渊, 等. 清香核桃在四川的引种表现及栽培技术 [J]. 中国南方果树, 2016, 45(2): 156–160. doi: 10.13938/j.issn. 1007-1431.20150532.
[17] FENG Y C, YANG S X, DONG S G, et al. An investigation of the introduction offrom Xinjiang in Lixian County in the arid valleys of the upper Minjiang River [J]. J Sichuan For Sci Technol, 2010, 31(3): 110–112. doi: 10.3969/j.issn.1003-5508.2010.03.020.冯云超, 杨素香, 董生刚, 等. 岷江上游干旱河谷理县新疆核桃引种调查 [J]. 四川林业科技, 2010, 31(3): 110–112. doi: 10.3969/j.issn. 1003-5508.2010.03.020.
[18] PAN J, ZHANG G S. Breeding report ofבShuxing 1’ and ‘Shuxing 6’ [J]. China For Ind, 2016(7): 290. 潘济, 张光胜. 蜀兴1号、6号核桃良种选育报告 [J]. 中国林业产业, 2016(7): 290.
[19] FANG W L, YANG Z B, HUANG Q, et al. Selective breeding of precocious, prolific and quality walnut strains [J]. Nonwood For Res, 1998(1): 6–10. 方文亮, 杨振帮, 黄谦, 等. 核桃早实、丰产、优质杂交新品系的选育研究[J]. 经济林研究, 1998(1): 6–10.
[20] ZHOU L Y, PU G L, XIAO Q G, Study on the photosynthetic and physiological hybrid vigor of walnut [J]. N Hort, 2009(10): 41–44. 周兰英, 蒲光兰, 肖前刚. 核桃杂交后代光合生理优势研究 [J]. 北方园艺, 2009(10): 41–44.
[21] PU G L, XIAO Q W, ZHOU L Y. A New early-fruiting walnut cultivar ‘Chuanzao 1’ [J]. Acta Hort Sin, 2011, 38(10): 2025–2026. doi: 10. 16420/j.issn.0513-353x.2011.10.025.蒲光兰, 肖千文, 周兰英. 早实核桃新品种‘川早1号’ [J]. 园艺学报,2011, 38(10): 2025–2026. doi: 10.16420/j.issn.0513-353x.2011.10.025.
[22] XIAO Q W, XIAO Q G, ZHOU L Y, et al. A new early-maturing and thin shell walnut cultivar ‘Shuangzao’ [J]. Acta Hort Sin, 2013, 40(1): 179–180. doi: 10.16420/j.issn.0513-353x.2013.01.020.肖千文, 肖前刚, 周兰英, 等. 早熟薄皮核桃新品种‘双早’ [J]. 园艺学报, 2013, 40(1): 179–180. doi: 10.16420/j.issn.0513-353x.2013.01. 020.
[23] BAIRD N A, ETTER P D, ATWOOD T S, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers [J]. PLoS One, 2008, 3(10): e3376. doi: 10.1371/journal.pone.0003376.
[24] LI H, DURBIN R. Fast and accurate short read alignment with Burrows- Wheeler transform [J]. Bioinformatics, 2009, 25(14): 1754–1760. doi: 10.1093/bioinformatics/btp324.
[25] McKENNA A, HANNA M, BANKS E, et al. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data [J]. Genome Res, 2014, 20(9): 1297–1303. doi: 10. 1101/gr.107524.110.
[26] LI H, HANDSAKER B, WYSOKER A, et al. Genome project data processing S: The sequence alignment/map format and samtools [J]. Bioinformatics, 2009, 25(16): 2078–2079. doi: 10.1093/bioinformatics/ btp352.
[27] STAMATAKIS A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies [J]. Bioinformatics, 2014, 30(9): 1312–1313. doi.org/10.1093/bioinformatics/btu033.
[28] FALUSH D, STEPHENS M, PRITCHARD J K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies [J]. Genetics, 2003, 164(4): 1567–1587. doi: 10.3410/ f.1015548.197423.
[29] YANG J, LEE S H, GODDARD M E, et al. GCTA: A tool for genome- wide complex trait analysis [J]. Amer J Hum Genet, 2011, 88(1): 76–82. doi: 10.1016/j.ajhg.2010.11.011.
[30] PFEIFER B, WITTELSBURGER U, RAMOS-ONSINS S E, et al. PopGenome: An efficient Swiss army knife for population genomic analyses in R [J]. Mol Biol Evol, 2014, 31(7): 1929–1936. doi: 10. 1093/molbev/msu136.
[31] Brown G R, Gill G P, Kuntz R J, et al. Nucleotide diversity and linkage disequilibrium in loblolly pine [J]. Proc Natl Acad Sci USA, 2004, 101(42): 15255–15260. doi: org/10.1073/pnas.0404231101.
[32] INGVARSSON P K. Nucleotide polymorphism and linkage dise- quilibrium within and among natural populations of European aspen (L., Salicaceae) [J]. Genetics, 2005, 169(2): 945–953. doi: org/10.1534/genetics.104.034959.
[33] POLLEGIONI P, WOESTE K E, CHIOCCHINI F, et al. Ancient humans influenced the current spatial genetic structure of common walnut populations in Asia [J]. PLoS One, 2015, 10(9): e0135980. doi: 10.1371/journal.pone.0135980.
[34] YAN Z F, SHANG X Y. Studies on classification of walnut in Xinjiang [J]. Xinjiang Agric Sci, 1987(5): 25–26. 严兆福, 尚新业. 新疆核桃分类的探讨 [J]. 新疆农业科学, 1987(5): 25–26.
[35] WANG H, PEI D, GU R S, et al. Genetic diversity and structure of walnut populations in central and southwestern China revealed by microsatellite markers [J]. J Amer Soc Hort Sci, 2008, 133(2): 197– 203.
[36] SUN Y W. History and resources of Chinese fruit tree [M]. Shanghai: Shanghai Scientific & Technical Publishers, 1983: 12–13. 孙云蔚. 中国果树史与果树资源 [M]. 上海: 上海科学技术出版社, 1983: 12–13.
[37] WANG S D, MAO G H, HU C L, et al. Grafting techniques of improving wild low-yield and low-quality walnut trees in Yanyuan County [J]. J Sichuan For Sci Technol, 2010, 31(3): 97–101,70. doi: 10. 3969/j.issn.1003-5508.2010.03.017.王仕娣, 毛国慧, 胡聪林, 等. 盐源县低产低质野生核桃改良嫁接技术试验[J]. 四川林业科技, 2010, 31(3): 97–101,70. doi: 10.3969/ j.issn.1003-5508.2010.03.017.
[38] SUN Q, XIAO Q W, LUO Y F, et al. Research on the main economic characters of the giant walnut in Shimian [J]. N Hort, 2011(18): 15–18. 孙权, 肖千文, 罗永飞, 等. ‘石棉巨型’核桃主要经济性状研究 [J]. 北方园艺, 2011(18): 15–18.
[39] WU C L, ZHANG Q Q, DONG B X, et al. Analysis of genetic structure and genetic relationships of partial maize inbred lines in China [J]. Acta Agric Sin, 2010, 36(11): 1820–1831. doi: 10.3724/SP. J.1006.2010.01820.吴承来, 张倩倩, 董炳雪, 等. 我国部分玉米自交系遗传关系和遗传结构解析 [J]. 作物学报, 2010, 36(11): 1820–1831. doi: 10.3724/ SP.J.1006.2010.01820.
[40] XU G B. Genetics of Plant Population [M]. Beijing: Science Press, 2009: 197-198. 徐刚标. 植物群体遗传学 [M]. 北京: 科学出版社, 2009: 197-198.
[41] WANG J Y, WU C F, TANG Y. Preliminary study on genetic diversity of(Magnoliaceae) through SNP marker [J]. Guihaia, 2012, 32(4): 542–547. doi: 10.3969/j.issn.1000-3142.2012. 04.022.王佳媛, 吴传芳, 唐亚. 基于SNP分子标记的凹叶木兰遗传多样性初步研究 [J]. 广西植物, 2012, 32(4): 542–547. doi: 10.3969/j.issn. 1000-3142.2012.04.022.
[42] WANG Y S. Research progress of plant population genomics based on high-throughput sequencing [J]. Hereditas (Beijing), 2016, 38(8): 688– 699. doi: 10.16288/j.yczz.16-061.王云生. 基于高通量测序的植物群体基因组学研究进展 [J]. 遗传, 2016, 38(8): 688–699. doi: 10.16288/j.yczz.16-061.
[43] NORDBORG M, HU T T, ISHINO Y, et al. The pattern of polymer- phism in[J]. PLoS Biol, 2005, 3(7): 1289–1299. doi: 10.1371/journal.pbio.0030196.
[44] CAICEDO A L, WILLIAMSON S H, HERNANDEZ R D, et al. Genome- wide patterns of nucleotide polymorphism in domesticated rice [J]. PLoS Genet, 2007, 3(9): 1745–1756. doi:10.1371/journal.pgen.003 0163.
[45] CHU Y G, SU X H. Research progress of single nucleotide polymer- phisms in forest trees [J]. Hereditas (Beijing), 2008, 30(10): 1272– 1278. doi: 10.3321/j.issn:0253-9772.2008.10.006.褚延广, 苏晓华. 单核苷酸多态性在林木中的研究进展 [J]. 遗传, 2008, 30(10): 1272–1278. doi: 10.3321/j.issn:0253-9772.2008.10.006.
[46] HEUERTZ M, de PAOLI E, KÄLLMAN T, et al. Multilocus patterns of nucleotide diversity, linkage disequilibrium and demographic history of Norway spruce [(L.) Karst] [J]. Genetics, 2006, 174(4): 2095–2105. doi: 10.1534/genetics.106.065102.
[47] TUSKAN G A, DIFAZIO S, JANSSON S, et al. The genome of black cottonwood,(Torr. & Gray) [J]. Science, 2006, 313(5793): 1596–1604. doi: 10.1126/science.1128691.
[48] CAMPOY J A, LERIGOLEUR-BALSEMIN E, CHRISTMANN H, et al. Genetic diversity, linkage disequilibrium, population structure and construction of a core collection ofL. landraces and bred cultivars [J]. BMC Plant Biol, 2016, 16: 49. doi: 10.1186/s12870-016- 0712-9.
[49] EMANUELLI F, LORENZI S, GRZESKOWIAK L, et al. Genetic diversity and population structure assessed by SSR and SNP markers in a large germplasm collection of grape [J]. BMC Plant Biol, 2013, 13: 39. doi: 10.1186/1471-2229-13-39.
[50] HAN H, WOESTE K E, HU Y H, et al. Genetic diversity and population structure of common walnut () in China based on EST-SSRs and the nuclear gene phenylalanine ammonia-lyase () [J]. Tree Genet Gen, 2016, 12(6): 111. doi:10.1007/s11295-016-1064-1.
[51] WU T, XIAO L J, CHEN S Y, et al. Transcriptomics and comparative analysis of threespecies;,and[J]. Plant Omics, 2015, 8(4): 361–371.
[52] NIELSEN R. Molecular signatures of natural selection [J]. Ann Rev Genet, 2005, 39: 197–218. doi: 10.1146/annurev.genet.39.073003.112 420.
Studies on Genetic Diversity ofCultivar Germplasms in Sichuan Based on RAD-SNPs Analysis
YAN Si-yu1*, ZHU Peng1*, GONG Wei1**, WANG Jing-yan1, WU Kai-zhi2, WU Chun-yan2, LI Hai-ping2, GONG Yi-hong2, DUAN Qiong2
(1. College of Forestry, Sichuan Agricultural University, Chengdu 611130, China; 2. Forest Seedling Station of Sichuan Province,Chengdu 610081, China)
In order to understand the genetic diversity of walnut () in Sichuan, the genetic diversity and genetic structure of 42 walnut germplasms were analyzed by RAD-SNPs method. The results showed that a total of 70G clean data and 160 309 high consistent SNPs were obtained, and the average Q30 was 96.3%. Forty-two walnut genotypes could be divided into two main groups, such as AJR (group,=33) and AJS (group,=9). The high genetic differentiation (ST=0.285) between two groups had correlation with their geographic distribution. Meanwhile, the groups of three varieties of ‘Weibo No. 1’, ‘Bailong No. 1’ and ‘Shimianjuhe’ were adjusted. There were 9 and 14 varieties in AJS and AJR groups with pure blood, respectively, while the rests in AJS and AJR groups were mixed blood. There were high genetic diversity among 42 walnut germplasms with nucleotide diversity [P()] of0.029and expected heterozygosity () of0.286, and thegroup has higher genetic diversity thangroup. So, these would provide basis for the preservation and cross breeding of walnut germplasm resources in Sichuan.
RAD-SNP;;; Genetic diversity
10.11926/jtsb.3906
2018-03-15
2018-05-09
四川省林业厅种苗站项目(20140312);四川省科技厅项目(2016NYZ0035, 2017NFP0051, 2017NFP0126);四川省农业科技成果转化项目(16NZ0067); 四川农业大学新农村发展研究院雅安服务总站项目(2017-05)资助
This work was supported by the Projects of Tree Seedling Station of Sichuan Forestry Department (Grant No. 20140312), and the Projects of Science and Technology Department of Sichuan Province (Grant No. 2016NYZ0035, 2017NFP0051, 2017NFP0126), the Project for Agricultural Science and Technology Achievements Transformation in Sichuan (Grant No. 16NZ0067), and the Project of Yaan Service Station of the Institute for New Rural Development, Sichuan Agriculture University (Grant No. 2017-05).
闫思宇(1993~ ), 女, 硕士研究生, 主要从事经济林方面研究。E-mail: ysy4017@126.com
* 对本文有同等贡献,并列第一作者
E-mail: gongwei@sicau.edu.cn
