基于SNP标记的菜豆品种真实性和纯度鉴定技术
2019-02-10颜廷进蒲艳艳张文兰田茜王栋李群戴双丁汉凤
颜廷进 蒲艳艳 张文兰 田茜 王栋 李群 戴双 丁汉凤


摘要:为了探索SNP标记技术在菜豆真实性和品种纯度鉴定中的应用,采用2b-RAD技术对200份国内外菜豆品种进行全基因组扫描、分析和评价,从中筛选出3 302个高分辨率、单拷贝和分布比较均匀的SNP标记,可以将除2个近等基因材料以外的198份品种资源区分开。基于组合最优化算法,利用 Haploview 4.2软件获得了菜豆资源鉴定最少SNP位点组合一套,包含46个 Tag- SNP标记,与3 302个SNP标记区分结果相同。本研究可为菜豆真实性和品种纯度鉴定技术体系的建立和指纹数据库的构建奠定基础。
关键词:菜豆;SNP标记;品种真实性;品种纯度;鉴定
中图分类号:S643.1 文献标识号:A 文章编号:1001-4942(2019)12-0111-09
Abstract In order to explore single nucleotide polymorphism (SNP) markers applying in authenticity and purity identification of common bean varieties, two hundred common bean varieties and landraces from China and abroad were conducted genome-wide scanning, analysed and evaluated by 2b-RAD technology. Three thousand three hundred and two SNP markers were screened out with high resolution, single copy and uniform distribution in the linkage map, which could distinguish all the varieties and landraces except two near-isogenic materials. Based on the combinatorial optimization algorithm, 46 Tag-SNP markers as the minimum site combination of SNPs were obtained by Haploview 4.2 software,Whose distinguishing ability was as same as that of the 3 302 SNP markers. The study provided a foundation for the identification of variety genuineness and purity and the construction of fingerprint database of common bean.
Keywords Common bean; SNP markers; Variety genuineness; Variety purity; Identification
菜豆(Phaseolus vulgaris L.)作为世界范围内重要的食用豆类作物,拥有最大的豆类种植面积[1],产量约占全球食用豆的一半[2]。它起源于中美洲和安第斯山两个中心[3],并于500多年前引入中国进行栽培[4]。我国菜豆品种资源较为丰富[5],是世界上普通菜豆生产和消费的主产区之一。作为我国重要的植物蛋白质来源,菜豆在贫困地区营养供应和人类繁衍发展方面发挥了重要作用,同时也为发达地区膳食结构的调整做出巨大贡献[6]。
农作物品种真实性鉴定和纯度分析是农业生产发展过程中的重要技術和关键环节。随着作物生物学研究的发展,菜豆品种的真实性和纯度技术分析经历了多个阶段。形态学方法主要是田间小区种植鉴定,依靠品种间的特征特性差异进行品种区分,是评价作物真实性、纯度鉴定和遗传多样性的有效手段之一[7-9]。较多研究证实利用形态标记对普通菜豆品种进行真实性鉴定、结构分析和多样性研究非常有效,有助于进一步了解品种形态变异特点及亚群之间的关系[10-13],但易受人为因素、环境气候条件的影响,无法保证检测结果的准确性及时效性。
随着分子群体遗传学理论和技术的发展,朊蛋白、同工酶等被用于作物品种结构和遗传多样性研究。其中朊蛋白标记方法在普通菜豆遗传结构及多样性研究中应用最广泛[14-17]。蛋白质和同工酶是基因表达的产物,其遗传方式并非全部表现为共显性遗传,多态性较少,对亲缘关系较近及遗传基础复杂的材料难以鉴别。近年来,DNA分子标记技术由于具有丰富的多态性和显著的个体特异性,能够在分子水平上识别品种间差异,不易受外界环境影响,可快速、准确地完成品种鉴定[18-20]。SSR分子标记技术已应用于遗传连锁图谱构建、品种鉴定和纯度分析、基因定位、图位克隆等多项研究中[21-24],同样应用于玉米、小麦、甘蓝、大豆、水稻、棉花、马铃薯等作物的鉴定工作[25-31]。但在实际应用过程中,SSR标记出现了不易实现数据整合和通量低等缺陷。单核苷酸多态性(single nucleotide polymorphism,SNP)标记作为最新发展起来的分子标记,具有检测通量大、分布密度广、多态性高、遗传稳定性强、速度快等特点,并且国内外很多公司推出的各种SNP分析检测平台,其分析系统自动化程度高、易于标准化操作,很好地弥补了SSR标记的技术缺陷,适合大规模SNP研究及基因分型。迄今,SNP标记技术已在水稻[32]、玉米[33,34]、大豆[35]、大麦[36]、棉花[37]等作物品种鉴定研究中得到广泛应用。
本研究将SNP标记技术应用于200份普通菜豆的品种鉴定,利用抽提得到的DNA,采用2b-RAD技术,使用标准型5′-NNN-3′接头与酶切标签连接,于文库质控合格后在Illumina Hiseq Xten平台进行双端测序,以期筛选出鉴定不同菜豆品种资源的最少SNP位点组合,并为菜豆品种测序真实性和纯度鉴定技术体系的建立和指纹数据库的构建奠定基础。
1 材料与方法
1.1 试验材料
供试样品为200份菜豆品种(表1)。其中,152份材料引自美国西部植物引种站(Western Regional Plant Introduction Station),来源于墨西哥、哥伦比亚、秘鲁、危地马拉、玻利维亚等10个国家;48份为从国家资源库引进的国内品种,材料类型分为地方品种和栽培品种。
1.2 DNA提取和2b-RAD测序
采用Ezup柱式植物基因组DNA抽提试剂盒[生工生物工程(上海)股份有限公司产品]抽提菜豆基因组DNA,具体操作方法参照试剂盒说明书,并通过1%琼脂糖凝胶电泳和Thermo Scientific NanoDrop ND-2000进行DNA质量和浓度检测。
2b-RAD是一种简化基因组测序技术,其原理是使用ⅡB型限制性核酸内切酶将基因组DNA进行酶切,然后对酶切片段进行测序。本试验所提DNA经检测后利用该技术进行酶切,酶切后的简化基因组测定由青岛欧易生物科技有限公司Illumina Hiseq Xten平台完成。
1.3 测序质量分析和SNP基因分型
利用Illumina Hiseq Xten平台对2b-RAD技术构建的200个菜豆样品标签测序文库进行双端(paired-end)测序,采用标准型5′-NNN-3′接头建库。将原始reads(测序时的最小序列单位)按照以下条件进行过滤:(1)剔除不含有限制性内切酶(BsaXⅠ)识别位点的序列;(2)剔除低质量序列(质量值低于30的碱基超过15%,判定为低质量reads);(3)剔除含N碱基的序列。将得到的高质量reads用于后续分析。
对于有参考基因组的标记,采用RAD typing软件,利用最大似然法进行分型。为了保证SNP位点分型的准确性和严谨性,进行以下条件的过滤:(1)剔除最小的等位基因频率(MAF)小于0.01的SNP位点;(2)剔除SNP分型缺失率高于20%的位点;(3)剔除同一个标签上SNP位点多于2个的数据。
1.4 数据分析
使用Haploview 4.2软件进行SNP标签统计分析,从而获得鉴定菜豆所用的最少SNP位点组合,即核心SNP位点;通过TreeBeST 1.9.2软件计算距离矩阵,构建系统进化树。
2 结果与分析
2.1 SNP位点分布
参照Fu[38]等的基因分型策略,以菜豆为参考基因组(ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/499/845/GCF_000499845.1_PhaVulg1_0/GCF_000499845.1_PhaVulg1_0_genomic.fna.gz),根据比对结果统计样品获得的标签数及深度信息并计算测序数据对参考基因组的覆盖度。200个样品的标签平均数目为83 497,平均测序深度为29×,覆盖度范围0.38%~0.49%;获得25 561个SNP位点,使用划窗口的方式绘制SNP在染色体上的分布图,窗口大小设置为200 kbp,每次移动100 kbp,统计区间内的SNP数目,结果如图1所示。SNP在菜豆染色体上的平均分布密度为每kb 4.92×10-2个。
2.2 核心SNP位点筛选
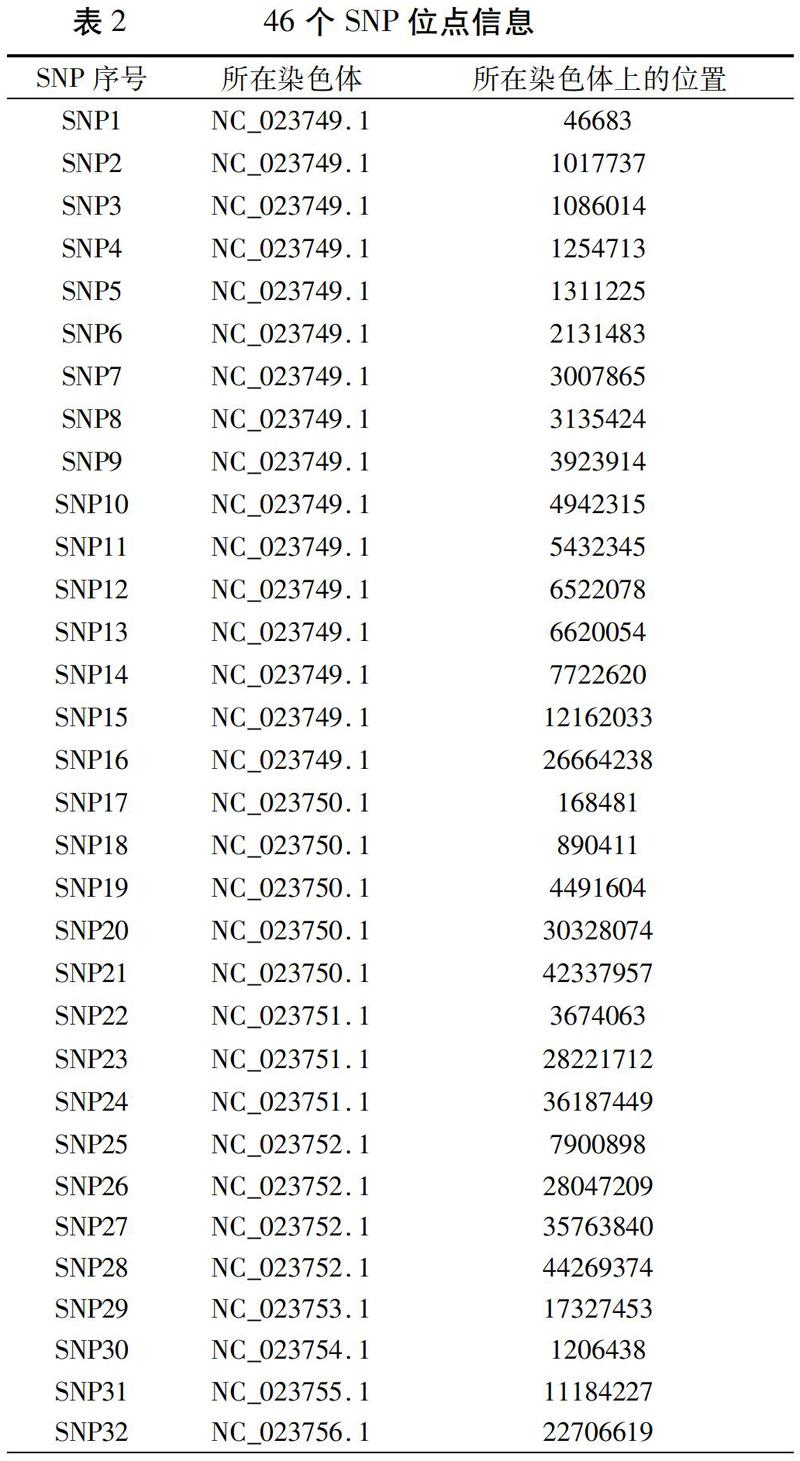
从获得的25 561个SNP位点中删除有缺失的SNP,剩余3 302个SNP位点,再用 Haploview 4.2软件挑选 tag SNP,筛选条件: MAF=0.2,哈迪温伯格平衡 p-value>10-7。试验共筛选得到19个位点,将这19个位点连接成序列,得到146条序列。其中有四条序列分别对应16、17、5、3个菜豆样品。用 Haploview 软件继续挑选16个样品中的tag SNP,结合人工筛选,挑选出10个位点,可以将该类样品分开。利用同样的方法挑选tag SNP区分包含17、5、3个等个数的样品,共挑选出17个位点。综合分析可得,使用46个位点可以将198个品种样品区分开来(表2)。
2.3 品种鉴别方法
将每个样品的3 302个没有缺失的位点连接成序列后,得到199条不同序列,可以很容易区分198个菜豆品种。为了进一步简化操作程序,节约试验成本,通过软件分析结合人工挑选,筛选出46个核心位点。将每个品种的46个核心位点按染色体排列顺序从左到右、从上到下连接成序列,也获得了不同的199条序列(表3)。利用每个品种之间的位点差异、排列顺序的不同,同样可达到区分198个菜豆品种的目的。由于P-190 和 P-200 两个样品亲缘关系较近,不能用此进行区分,因而必须使用有缺失的SNP位点(NC_023749.1-7722596位点)。本试验共通过47个SNP位点完成了200个菜豆品种的区分工作。
2.4 系统进化树
将每个SNP标记首尾相连,如果缺失相应的位点,则用“-”代替。获得的序列采用邻接法(neighbor-joining method)构建进化树,利用TreeBesT 1.9.2软件计算距离矩阵,进化树的可靠性通过bootstrap 法进行检验(重复1 000次)。由图2可以很清晰地看到,本试验建立的SNP标记法可以将200份菜豆品种按照亲缘关系的远近分类,从而进一步探明了200份菜豆的进化历程和亲缘关系。
3 讨论与结论
快速、准确、经济的检测技术是研究工作者的追求目标。宋伟等[39]利用42个SNP位点的基因分型数据信息成功区分105份玉米自交系材料;刘丽华等[40]研究表明14个SNP标记与384个SNP标记区分能力相同,可区分378个小麦材料中的376个,仅有两个近等位基因系的品种无法区分,区分开的比例为99.5%。本研究利用46個SNP位点成功对198个菜豆品种进行区分,区分开的比例为99%,对于亲缘关系特别近的两个样品P1-199和P-200采用SNP缺失的办法进行了区分。但随着菜豆新品种数量的增加,已筛选出的核心SNP位点能否满足要求,仍需进一步验证,并且还可能需要再确定新的核心位点。但对于亲缘关系特别近的品种,即使再增加核心位点也无法进行区分,建议使用“核心位点+扩展位点”相结合的模式,达到完全区分所有品种的目的。
目前,Cavanagh等[41]和Wang等[42]分别开发了小麦9k和90k高通量SNP分析芯片,用于小麦身份鉴定。以后的应用研究中,我们应进一步验证菜豆46个核心SNP位点的有效性,若能成功区分所有菜豆品种,可以考虑将46个SNP位点连成序列,做成芯片,进而构建菜豆品种真实性和品种鉴定数据库,直接用于菜豆品种真实性和品种纯度鉴定。
参 考 文 献:
[1] Kole C. Wild crop relatives: genomic and breeding resources legume crops and forages[M]. New York: Springer, 2011.
[2] Wilson R F, Stalker H T, Brummer E C. Legume crop genomics[M]. Champaign: AOCS Press, 2004: 61-82.
[3] 张晓艳, 王坤, 王述民. 普通菜豆种质资源遗传多样性研究进展[J]. 植物遗传资源学报, 2007,8(3): 359-365.
[4] 郑卓杰. 中国食用豆类学[M]. 北京: 中国农业出版社, 1997.
[5] 郝晓鹏, 田翔, 王燕, 等. 山西普通菜豆种质资源籽粒品质分析和评价[J]. 山西农业科学, 2016, 44(6): 808-810,832.
[6] 宗绪晓. 食用豆类高产栽培与食品加工[M]. 北京: 中国农业科学技术出版社, 2002: 227-249.
[7] Newbury H J, Ford-Lloyd B V. Estimation of genetic diversity[M]. Dordrecht: Springer, 2000: 192-206.
[8] Delacy I H,Skovmand B,Huerta J. Characterization of Mexican wheat landraces using agronomically useful attributes[J]. Genetic Resources and Crop Evolution, 2000, 47(6): 591-602.
[9] Dotlacil L, Hermuth J, Stehno Z, et al. Diversity in European winter wheat landraces and obsolete cultivars[J]. Czech Journal of Genetics & Plant Breeding, 2000, 36: 29-36.
[10]欒非时, 崔成焕, 王金陵. 菜豆种质资源形态标记的研究[J]. 东北农业大学学报, 2001, 32(2): 134-138.
[11]张赤红, 曹永生, 宗绪晓, 等. 普通菜豆种质资源形态多样性鉴定与分类研究[J]. 中国农业科学, 2005, 38(1): 27-32.
[12]张晓艳, 王坤, Blair M W, 等. 中国普通菜豆形态性状分析及分类[J]. 植物遗传资源学报, 2007, 8(4): 406-410.
[13]王兰芬, 武晶, 王昭礼, 等. 普通菜豆种质资源表型鉴定及多样性分析[J]. 植物遗传资源学报, 2016, 17(6):976-983.
[14]Igrejas G, Carnide V, Pereira P, et al. Genetic diversity and phaseolin variation in Portuguese common bean landraces[J]. Plant Genetic Resources, 2009, 7(3): 230-236.
[15]雷蕾, 王兰芬, 武晶, 等. 中国普通菜豆种质资源朊蛋白变异及多样性分析[J]. 植物遗传资源学报, 2017, 18(6): 1006-1012.
[16]Negahi A, Bihamta M R, Negahi Z. Diversity in Iranian and exotic common bean (Phaseolus vulgaris L.) using seed storage protein (phaseolin)[J]. Agricultural Communications, 2014, 2(4): 34-40.
[17]Carovic'-Stanko K, Liber Z, Vidak M, et al. Genetic diversity of Croatian common bean landraces[J]. Frontiers in Plant Science, 2017, 8: 604.
[18]Roy S, Ray B P, Sarker A, et al. DNA fingerprinting and genetic diversity in lentil germplasm using SSR markers[J]. Asian Journal of Conservation Biology, 2015, 4(2): 109-115.
[19]Brown S, Higham T, Slon V, et al. Identification of a new hominin bone from Denisova Cave, Siberia using collagen finger printing and mitochondrial DNA analysis[J]. Scientific Reports, 2016, 6:23559.
[20]Salimizand H, Menbari S, Ramazanzadeh R, et al. DNA fingerprinting and antimicrobial susceptibility pattern of clinical and environmental Acinetobacter baumannii isolates: a multicentre study[J]. Journal of Chemotherapy, 2016, 28(4): 277-283.
[21]张增翠, 侯喜林. SSR分子标记开发策略及评价[J]. 遗传, 2004, 26(5): 763-768.
[22]Min S K, Choi B, Park J H, et al. Assessment of genetic diversity and population structure of the sub core set in sesame(Sesamum indicum) using SSR markers[J]. Gene, 2016, 28(1): 73-83.
[23]Dencic S, Depauw R, Momcilovic V, et al. Comparison of similarity coefficients used for cluster analysis based on SSR markers in sister line wheat cultivars[J]. Genetika, 2016, 48(1): 219-232.
[24]An M, Deng M, Zheng S S , et al. De novo transcriptome assembly and development of SSR markers of oaks Quercus austrocochinchinensis and Q. kerrii(Fagaceae)[J]. Tree Genetics & Genomes, 2016, 12(6): 103-112.
[25]王鳳格, 杨扬, 易红梅, 等. 中国玉米审定品种标准SSR 指纹库的构建[J]. 中国农业科学, 2017, 50(1): 1-14.
[26]Yumiko F, Hiroyuki F, Hiroshi Y. Identification of wheat cultivars using EST-SSR markers[J]. Breeding Science, 2009, 59: 159-167.
[27]王庆彪, 张扬勇, 庄木, 等. 中国50个甘蓝代表品种EST-SSR指纹图谱的构建[J]. 中国农业科学, 2014, 47(1): 111-121.
[28]谢华, 关荣霞, 常汝镇, 等. 利用SSR标记揭示我国夏大豆(Glycine max(L.) Merr)种质遗传多样性[J]. 科学通报, 2005, 52(5): 343-351.
[29]张云, 龙云星, 奎丽梅, 等. 我国水稻两用核不育系 SSR 遗传多样性分析[J]. 分子植物育种,2017, 15(7): 2836-2846.
[30]匡猛, 杨伟华, 许红霞, 等. 中国棉花主栽品种 DNA 指纹图谱构建及SSR标记遗传多样性分析[J]. 中国农业科学, 2011, 44(1): 20-27.
[31]段艳凤, 刘杰, 卞春松, 等. 中国88个马铃薯审定品种SSR指纹图谱构建与遗传多样性分析[J]. 作物学报, 2009, 35(8): 1451-1457.
[32]Shirasawa K, Shiokai S, Yamaguchi M, et al. Dot-blot-SNP analysis for practical plant breeding and cultivar identification in rice[J]. Theoretical and Applied Genetics, 2006, 113(1):147-155.
[33]Jones E S, Sullivan H, Bhattramakkid A, et al.A comparison of simple sequence repeat and single nucleotide polymorphismmarker technologies for the genotypic analysis of maize(Zea mays L.)[J]. Theoretical and Applied Genetics, 2007, 115(3): 361-371.
[34]Tian H L, Wang F G, Zhao J R, et al. Development of maize SNP3072, a high-throughput compatible SNP array, for DNA fingerprinting identification of Chinese maize varieties[J]. Molecular Breeding, 2015, 35: 136.
[35]Yoon M S, Song Q J, Choii Y, et al. Barcsoy SNP23: a panel of 23 selected SNPs for soybean cultivar identification[J]. Theoretical and Applied Genetics, 2007, 114(5): 885-899.
[36]张利莎, 董国清, 扎桑, 等. 基于 EST-SSR和 SNP 标记的大麦麦芽纯度检测[J]. 作物学报, 2015, 41(8) :1147-1154.
[37]Kuang M, Wei S J, Wang Y Q, et al. Development of a core set of SNP markers for the identification of upland cotton cultivars in China[J]. Journal of Integrative Agriculture, 2016, 15(5): 954-962.
[38]Fu X, Dou J, Mao J, et al. RAD typing: an integrated package for accurate de novo codominant and dominant RAD genotyping in mapping populations[J]. PloS ONE, 2013, 8(11):e79960.
[39]宋偉, 王凤格, 田红丽, 等. 利用核心SNP位点鉴别玉米自交系的研究[J]. 玉米科学, 2013,21(4): 28-32.
[40]刘丽华, 庞斌双, 刘阳娜, 等. 基于 SNP 标记的小麦高通量身份鉴定模式[J]. 麦类作物学报,2018, 38(5): 529-534.
[41]Cavanagh C R, Chao S, Wang S, et al. Genome-wide comparative diversity uncovers multiple targets of selection for improvement in hexaploid wheat landraces and cultivars[J]. Proc. Natl. Acad. Sci. U. S. A., 2013, 110(20): 8057-8062.
[42]Wang S, Meyer E, Mckay J K, et al. 2b-RAD: a simple and flexible method for genome-wide genotyping[J]. Nature Methods, 2012, 9(8):808-810.
