Six-long non-coding RNA signature predicts recurrence-free survival in hepatocellular carcinoma
2019-01-29JingXianGuXingZhangRunChenMiaoXiaoHongXiangYuNongFuJingYaoZhangChangLiuKaiQu
Jing-Xian Gu, Xing Zhang, Run-Chen Miao, Xiao-Hong Xiang, Yu-Nong Fu, Jing-Yao Zhang, Chang Liu, Kai Qu
Abstract BACKGROUND Recent evidence shows that long non-coding RNAs (lncRNAs) are closely related to hepatogenesis and a few aggressive features of hepatocellular carcinoma(HCC). Increasing studies demonstrate that lncRNAs are potential prognostic factors for HCC. Moreover, several studies reported the combination of lncRNAs for predicting the overall survival (OS) of HCC, but the results varied. Thus,more effort including more accurate statistical approaches is needed for exploring the prognostic value of lncRNAs in HCC.AIM To develop a robust lncRNA signature associated with HCC recurrence to improve prognosis prediction of HCC.METHODS Univariate COX regression analysis was performed to screen the lncRNAs significantly associated with recurrence-free survival (RFS) of HCC in GSE76427 for the least absolute shrinkage and selection operator (LASSO) modelling. The established lncRNA signature was validated and developed in The Cancer Genome Atlas (TCGA) series using Kaplan-Meier curves. The expression values of the identified lncRNAs were compared between the tumor and non-tumor tissues. Pathway enrichment of these lncRNAs was conducted based on the significantly co-expressed genes. A prognostic nomogram combining the lncRNA signature and clinical characteristics was constructed.RESULTS The lncRNA signature consisted of six lncRNAs: MSC-AS1, POLR2J4, EIF3J-AS1,SERHL, RMST, and PVT1. This risk model was significantly associated with the RFS of HCC in the TCGA cohort with a hazard ratio (HR) being 1.807 (95%CI[confidence interval]: 1.329-2.457) and log-rank P-value being less than 0.001. The best candidates of the six-lncRNA signature were younger male patients with HBV infection in relatively early tumor-stage and better physical condition but with higher preoperative alpha-fetoprotein. All the lncRNAs were significantly upregulated in tumor samples compared to non-tumor samples (P < 0.05). The most significantly enriched pathways of the lncRNAs were TGF-β signaling pathway, cellular apoptosis-associated pathways, etc. The nomogram showed great utility of the lncRNA signature in HCC recurrence risk stratification.CONCLUSION We have constructed a six-lncRNA signature for prognosis prediction of HCC.This risk model provides new clinical evidence for the accurate diagnosis and targeted treatment of HCC.
Key words: Long non-coding RNAs; Hepatocellular carcinoma; Prognostic signature;Recurrence-free survival; Least absolute shrinkage and selection operator
INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth leading cause of cancer-related death worldwide and its incidence rate per year remains increasing rapidly[1]. HCC is an extremely heterogenous tumor from either clinical or molecular aspect, which is mainly due to the unique somatic genomic alteration patterns of each tumor[2]. In recent years, more mutation genes have been revealed to be involved in the tumorigenesis and progression of HCC, such as TP53, CTNNB1, and mTOR[3]. These molecular markers will help identify high-risk patients and provide guidance for therapeutic strategies directed at the individual patient. Due to the high heterogeneity of HCC, although therapeutic modalities have largely improved over the past decades, the prognosis remains unsatisfactory[4,5]. Therefore, more effective biomarkers for early diagnosis and precise prognostic prediction are in urgent need.
Long non-coding RNAs (lncRNAs) belong to the family of non-coding RNAs and measures longer than 200 nucleotides in length[6]. Accumulating evidence has shown that lncRNAs play an important part in a series of cellular biological processes and are associated with the initiation, progression, and migration of a wide range of malignancies including HCC[7]. However, so far, only a few lncRNAs like HOTAIR,HULC, and TERC have been well described about their oncogenic roles in HCC[8-10].Apart from them, more lncRNAs have been proposed as the diagnostic or prognostic biomarkers of HCC recently[11]. Yet by far, only a few prognostic models of HCC based on lncRNAs have been developed. For all we know, a total of three studies have reported the prognostic models based on lncRNAs of HCC[12-14]. Although all three previous studies used the overall group or part of The Cancer Genome Atlas (TCGA)cohort as the discovery dataset, the results varied. Therefore, to derive a more convincing result and find more potentially functional lncRNAs in HCC, in the present study, we selected GSE76427 from Gene Expression Omnibus (GEO) database as the discovery dataset and another independent dataset from TCGA database as the validation series. Moreover, the risk score system of HCC was formulated by a contemporary clinico-practical statistical method, the least absolute shrinkage and selection operator (LASSO) algorithm which was more accurate than multivariate COX model used by the previous three studies[15]. We here aimed to construct a robust lncRNA expression-based signature to improve prognostic prediction of HCC via comprehensive genomic data analysis.
MATERIALS AND METHODS
Microarray datasets
Microarray data of GSE76427 were downloaded from the Gene Expression Omnibus(https://www.ncbi.nlm.nih.gov/geo/) database. GSE764267 was conducted by GPL10558 (Illumina HumanHT-12 V4.0 expression beadchip). It contained 115 HCC tissue samples and 52 adjacent non-tumor tissue samples. Out of 115 tumor samples,108 with complete follow-up information (recurrence status and recurrence-free survival [RFS]) were included in the discovery dataset. All 108 participants underwent curative resection for HCC. Among the 108 participants, 22 were female and the other 86 were male. The average age of all the participants in the discovery dataset was 63.4 years old. With respect to HCC stage, 86 of 108 were in stage I or II,21 in stage III/IV, and only 1 patient had no record of cancer stage. The median follow-up was 1.17 years. HCC recurrence was diagnosed according to the established criteria reached by International Working Party[16]. Each array from GPL10558 consisted of more than 47,000 probes corresponding to more than 31,000 annotated genes including coding and non-coding genes, microRNAs, rRNAs, and other short RNAs. We extracted all the long non-coding RNAs from GPL10558 for the preliminary screening of prognostic lncRNAs. Three hundred and thirty-seven HCC patients with recurrence information from The Cancer Genome Atlas (TCGA,http://cancergenome.nih.gov/) constituted the validation dataset. The lncRNA expression profiles, clinical characteristics, follow-up data, and genetic mutation information of the TCGA cohort were downloaded. Besides, the RNA-Seq data of the 49 paired non-tumor tissue samples from the TCGA database were also obtained. The median RFS of GSE76427 and TCGA series was 8.4 and 13.0 months, respectively.
Construction and confirmation of an lncRNA signature
Univariate COX regression analysis was performed to screen the prognostic lncRNAs.Then, LASSO was applied to the construction of an HCC prognostic signature with the screened lncRNAs[15]. LASSO statistical algorithm was conducted using “glmnet”package in the R software (version 3.4.0, https://www.r-project.org/)[17]. Based on the expression levels of each sample, LASSO identified the eligible lncRNAs for the risk system and generated the corresponding coefficients for each of them.
The risk scores of each sample from the discovery and validation groups were calculated according to the risk model. The respective medians of two groups were used as the cut-off value to divide patients into high-risk and low-risk groups.Kaplan-Meier curves were plotted to compare the RFS of high-risk and low-risk patients. Meanwhile, P-values and hazard ratio (HR) with 95% confidence interval(CI) were generated by log-rank tests. Stratified survival analysis was carried out to identify the best candidates for the prognostic signature. The overall group was divided into subgroups by their clinical characteristics. Kaplan-Meier analysis was performed in the subgroups using the same cut-off value as the overall group.Kaplan-Meier curves were plotted using GraphPad Prism software (version 7.0).
Expression levels of the identified lncRNAs in tumor and non-tumor tissues were compared using TCGA RNA-seq data. The receiver operating characteristic (ROC)curve was plotted with GraphPad Prism (version 7.0). The area under the ROC curve(AUC) for evaluating discriminatory ability was calculated as well. Besides, the distribution of high-risk or low-risk patients in early- and late-stage subgroups were also compared via Chi-square test.
Function prediction of the prognostic lncRNAs
Pearson correlation analyses were conducted between the identified lncRNAs and the protein-coding genes in TCGA dataset based on their expression levels. The correlation coefficient > 0.4 and P < 0.001 were considered significantly correlated.The significantly co-expressed mRNAs were thrown into a publicly available web tool, Enrichr, for BioCarta (http://cgap.nci.nih.gov/Pathways/BioCarta_Pathways)pathway enrichment[18]. The enriched BioCarta terms were sorted by rank based ranking, an algorithm assessing the deviation from the expected rank to the mean rank[19].
Statistical analysis
Univariate and multivariate COX regression analyses were carried out in TCGA dataset to identify the independent risk factors for the RFS of HCC patients. A composite nomogram predicting the RFS of HCC was established based on the independent factors using the “rms” package of R statistical software. The concordance index (C-index) was calculated to evaluate the discriminatory ability of the nomogram. And calibration curves were plotted to compare the predicted and actual probabilities of RFS. Each component of the nomogram gives points and the sum of them represents the total points a patient receives. All the participants were divided into different risk groups according to their total points. Kaplan-Meier analysis was utilized to compare the RFS of different risk groups. Statistical analyses were performed with SPSS 23.0 (SPSS, Chicago, IL), unless otherwise indicated. A P-value < 0.05 was considered statistically significant.
RESULTS
Construction of a risk score system associated with RFS in HCC
The flow chart of the study procedure is presented in Figure 1. All the lncRNAs in the discovery dataset (GES76427) were subjected to univariate COX analysis and those significantly associated with RFS (P < 0.05) were considered as prognostic ones for LASSO modelling. The risk score formula for RFS was calculated as follows: risk score= 0.021355462 × (expression value of MSC-AS1) + 0.018051929 × (expression value of POLR2J4) + 0.016385849 × (expression value of EIF3J-AS1) + 0.01340867 × (expression value of SERHL) + 0.012263937 × (expression value of RMST) + 0.007303891 ×(expression value of PVT1). From the formula, it is seen that these lncRNAs were all risk factors for HCC recurrence (coefficient > 0). And the value of their respective coefficients represented how much impact they had on the RFS prediction. It is obvious that MSC-AS1 had the most while PVT1 had the least impact. The risk model generates a risk score for each participant. Using the median of the risk scores of the whole discovery group, 9.100, as the cut-off value, 108 patients were classified as high-risk or low-risk ones (Figure 2A). The recurrence status, RFS period, and six lncRNAs' expression value of each patient are presented in Figure 2A as well. Kaplan-Meier curves showed that the low-risk group had significantly longer RFS than the high-risk group with a P-value from log-rank test of 0.024 (HR = 1.842, 95%CI: 1.026-3.309) (Figure 2B).
Validation and development of a prognostic signature in TCGA cohort
To confirm the predictive ability of the lncRNA-signature, validation analysis was carried out in a group of 337 HCC patients from the TCGA project. The whole validation group was divided into high-risk and low-risk groups accordingly in the discovery dataset. The median score (0.0357) of the whole validation group was adopted as the cut-off value. Survival analysis showed great performance of the risk model in stratifying high-risk and low-risk patients with a log-rank P-value being less than 0.001 (HR = 1.807, 95%CI: 1.329-2.457) (Figure 2C).
Stratified survival analysis in the validation series was conducted to further investigate the suitable patient group of the six-lncRNA signature. The cut-off value in the subgroups was consistent with that in the overall group (0.0357). The result of stratification analysis was shown in Table 1. Our risk score system was more applicable to the patients possessing the following characteristics: TNM stage I/II,male gender, younger than 60 years, Asian, with family history, hepatitis B virus(HBV) infection, alcohol consumption, ECOG = 0, and higher levels of preoperative serum albumin (ALB, >3,5g/dl) and alpha-fetoprotein (AFP, >20 ng/ml).
Differential expression of the identified lncRNAs in tumor and non-tumor tissues
To investigate the expression profile of the identified lncRNAs, we compared the expression values of the six lncRNAs between tumor and non-tumor samples in TCGA dataset. The results showed that all six lncRNAs were significantly upregulated in HCC presenting with remarkably significant P-values (P < 1.0 × 10-10)(Figure 3A-F). In addition, the ROC curve showed the great utility of the combined six-lncRNA signature in discriminating tumor from non-tumor tissues. The AUC was 0.932 (95%CI: 0.898-0.966) (Figure 3G). Furthermore, the distribution of high-risk and low-risk patients in different stages was also examined. Patients in late-stage (TNM stage III/IV) had a higher likelihood of being high-risk patients than those in earlystage (TNM stage I/II) (P < 0.05), implying that higher risk score was associated with relatively late HCC stage (Figure 4H).
Figure 1 Overall design of the present study. HCC: Hepatocellular carcinoma; RFS: Recurrence-free survival.
Functional enrichment analysis of the six lncRNAs
To further investigate the potential biological roles of the identified six lncRNAs,BioCarta pathways were enriched using the co-expressed protein-coding genes of these lncRNAs. A gene significantly correlating with at least one of the six lncRNAs(Pearson coefficient > 0.4 and P <0.001) was considered eligible for pathway enrichment. Top ten highly enriched pathways are shown in Figure 4. These coexpressed genes of the lncRNAs clustered most significantly in TGF-β signaling pathway, internal ribosome entry pathway, granzyme A mediated apoptosis, FAS signaling pathway, calcium signaling by HBx, p38/MAPK signaling pathway, etc.Most of them are classical and vital pathways involved in HCC initiation and progression.
Establishment of a nomogram predicting RFS in HCC patients
To develop a composite predictor for the RFS of HCC patients, we combined the sixlncRNA signature, clinicopathological characteristics, and TP53 mutation status together for the screening of the independent factors for RFS. The results from univariate and multivariate COX regression analyses showed that the identified independent risk factors for RFS included the six-lncRNA score, TNM stage, and ECOG [Eastern Cooperative Oncology Group] score (P < 0.05) (Table 2). The nomogram for RFS prediction was comprised of the three factors (Figure 5A). The C-index of the nomogram was 0.684 (95%CI: 0.635-0.733). The calibration curves for the probability of recurrence at 1 year and 3 years showed good agreement between the prediction from the nomogram and the actual observations (Figure 5B). Each patient got the total points according to the scoring of the nomogram. The tertiles of all the total points were used as the cut-off value (6.800 and 3.200) to divide the patients into high-, intermediate- and low-risk groups. The Kaplan-Meier analysis of the three risk subgroups indicated the great utility of the composite nomogram in discriminating HCC patients with good, intermediate, and poor prognosis (Figure 5C).
DISCUSSION
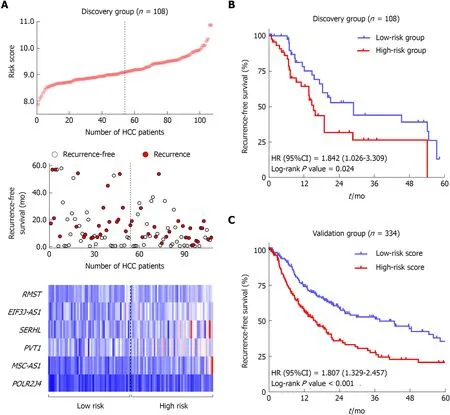
Figure 2 Construction and validation of a prognostic lncRNA signature for hepatocellular carcinoma. A: LncRNA risk score distribution (Upper), the recurrence status and recurrence-free survival (RFS) period (Middle), and expression profiles of the six lncRNAs (Lower) of the 108 patients in the discovery dataset. B: The Kaplan-Meier curve of the RFS between the high-risk and low-risk groups stratified by the median risk score in GSE76427 series. C: The Kaplan-Meier curve of the RFS between the high-risk and low-risk groups stratified by the median risk score in The Cancer Genome Atlas cohort.
Although extensive research efforts have been made on proposing the diagnostic and prognostic indicators of HCC in the past few decades, there is still a long way to go to construct a system of the molecular classifications of HCC[20]. A vast majority of the published studies investigating HCC predictors or predictive signatures were focused on protein-coding genes or microRNAs[21,22]. Currently, a group of non-coding RNAs,lncRNAs, which were overlooked previously, have attracted much attention. A growing number of studies have demonstrated that dysregulated lncRNAs in HCC are closely involved in the hepatocarcinogenesis, progression, and migration, and might be potential biomarkers for early detection, targeted therapies, and prognosis evaluation of HCC[7]. Thus, to help clarify the prognostic value of lncRNAs and refine prediction in HCC, we here carried out a comprehensive screening of the lncRNAs significantly associated with HCC recurrence in the public database (GSE76427) and used the significant lncRNAs to construct a prognostic model. We also presented the validation and development analysis of the established risk score system which showed good performance in predicting the RFS of HCC.
Compared to the lncRNA signatures of the three previous studies[12-14], our model was totally different. The reasons are probably as follows: First, our risk score system was constituted by LASSO penalized regression. Unlike traditional stepwise regression the previous studies used, LASSO algorithm can analyze all the independent variables simultaneously and tend to pick the most influential variables[17]. The coefficients of less influential variables will become zero after introduced to penalty following a regularization path[15]. Therefore, this formulation method was far more accurate than the stepwise regression of multivariate COX model, especially when dealing with very large datasets, like genomics[23]. Second, the three published studies all employed TCGA cohort as the discovery dataset while we used the GSE76427 for discovery and TCGA for validation. It was mainly thediscovery dataset that determined the key lncRNAs for further validation and investigation. Moreover, all the three studies adopted overall survival (OS) as an evaluating indicator of prognosis while our study used RFS for prognostic prediction,which implied that our signature might be more suitable for recurrence assessment.
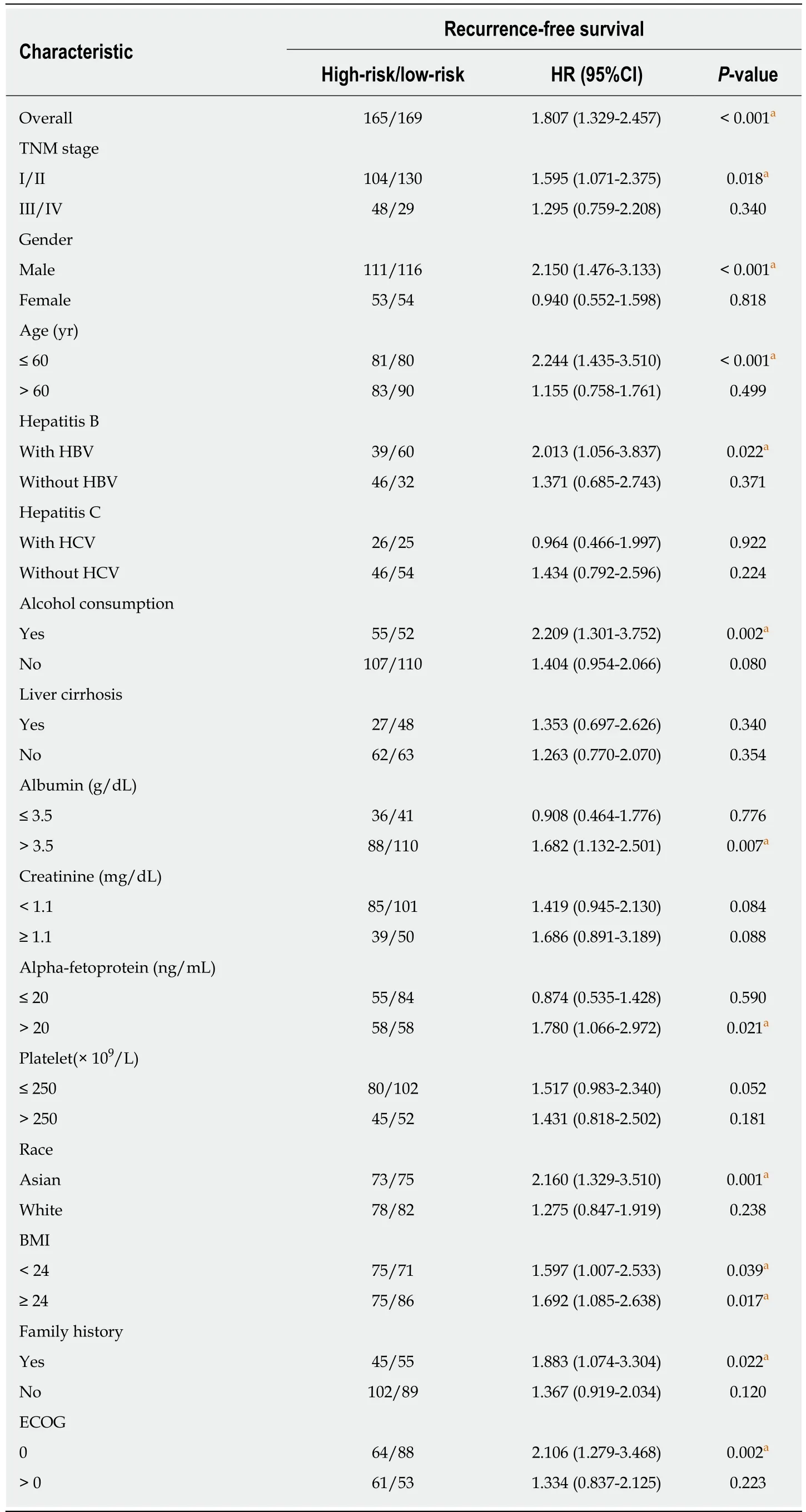
Table 1 Stratified analysis of recurrence-free survival in The Cancer Genome Atlas cohort
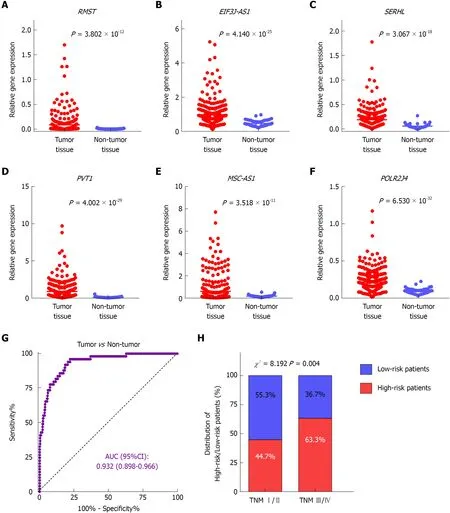
Figure 3 Expression patterns of the six lncRNAs in tumor and non-tumor tissues. Differential expression of RMST (A), EIF3J-AS1 (B), SERHL (C), PVT1 (D),MSC-AS1 (E), and POLR2J4 (F) between hepatocellular carcinoma and non-cancerous samples. G: The ROC curve of tumor tissue vs non-tumor tissue discriminated by the six-lncRNA signature. (H) Comparisons of the distribution of high-risk and low-risk patients in early stage (TNM I/II) and late stage (TNM III/IV) by the chi-square test.
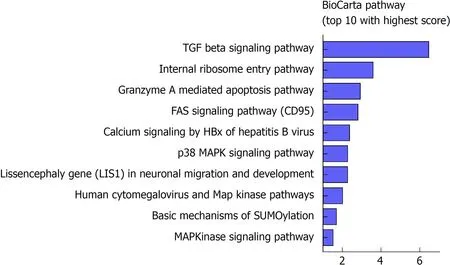
Figure 4 Functional annotation of the six lncRNAs. Top 10 significantly enriched BioCarta pathways using the coexpressed mRNAs of the six lncRNAs in The Cancer Genome Atlas database.
The expression values of the lncRNAs constituting the risk model in patients'tumor tissue can be tested via liver biopsy or from the surgical specimen. Out of the six lncRNAs, PVT1 (plasmacytoma variant translocation 1) has been demonstrated to be the activator of myelocytomatosis (MYC), a well-described oncogene[24]. PVT1 is upregulated in a wide variety of malignancies, particularly in digestive cancers, and is associated with a poor clinical outcome[25]. In HCC, studies showed that PVT1 could promote cell proliferation and stem-cell like potential by upregulating NOP2[26].Recent studies also revealed that PVT1 regulated the iron metabolism and cell apoptosis in HCC and promoted tumor progression and metastasis[27]. All these results suggested that PVT1 might be a powerful biomarker for HCC survival. Our prognostic signature enrolled PVT1, favoring the prognostic value of PVT1 in HCC as well. In case of RMST (rhabdomyosarcoma 2-associated transcript), it was reported to be involved in stem cell differentiation and neurogenesis[28]. However, to the best of our knowledge, there has not been any research into the role of RMST in HCC by now, either clinically or from the standpoint of molecular mechanism. Instead, our study provided novel evidence that RMST, as well as the other four lncRNAs (MSCAS1, POLR2J4, EIF3J-AS1, and SERHL), with no published studies reporting their biological functions so far as we know, might be potential predictors of HCC prognosis, and further studies are needed to validate these results and investigate its molecular characteristics.
In this study, we identified a novel and robust lncRNA signature for prognostic prediction of HCC. Stratified survival analysis showed that this six-lncRNA signature was more suitable for the recurrence prediction of relatively younger (aged ≤ 60 years) Asian male patients with HBV infection, family history, and history of alcohol consumption who are in TNM stage I or II and better physical condition (ECOG = 0 and preoperative ALB > 3.5 g/dL) but with higher preoperative AFP. To further refine prediction, a nomogram combining the molecular signature and clinical markers was constructed. Although the biological functions of the identified lncRNAs in HCC have not been researched or reported except PVT1, pathway enrichment revealed that these lncRNAs might exert influence on tumorigenesis and progression of HCC through the TGF-β pathway and cellular apoptosis-associated pathways. In a word, our study highlighted the prognostic value of the six-lncRNA signature and suggested practical applications in prognostic prediction and targeted therapy of HCC.
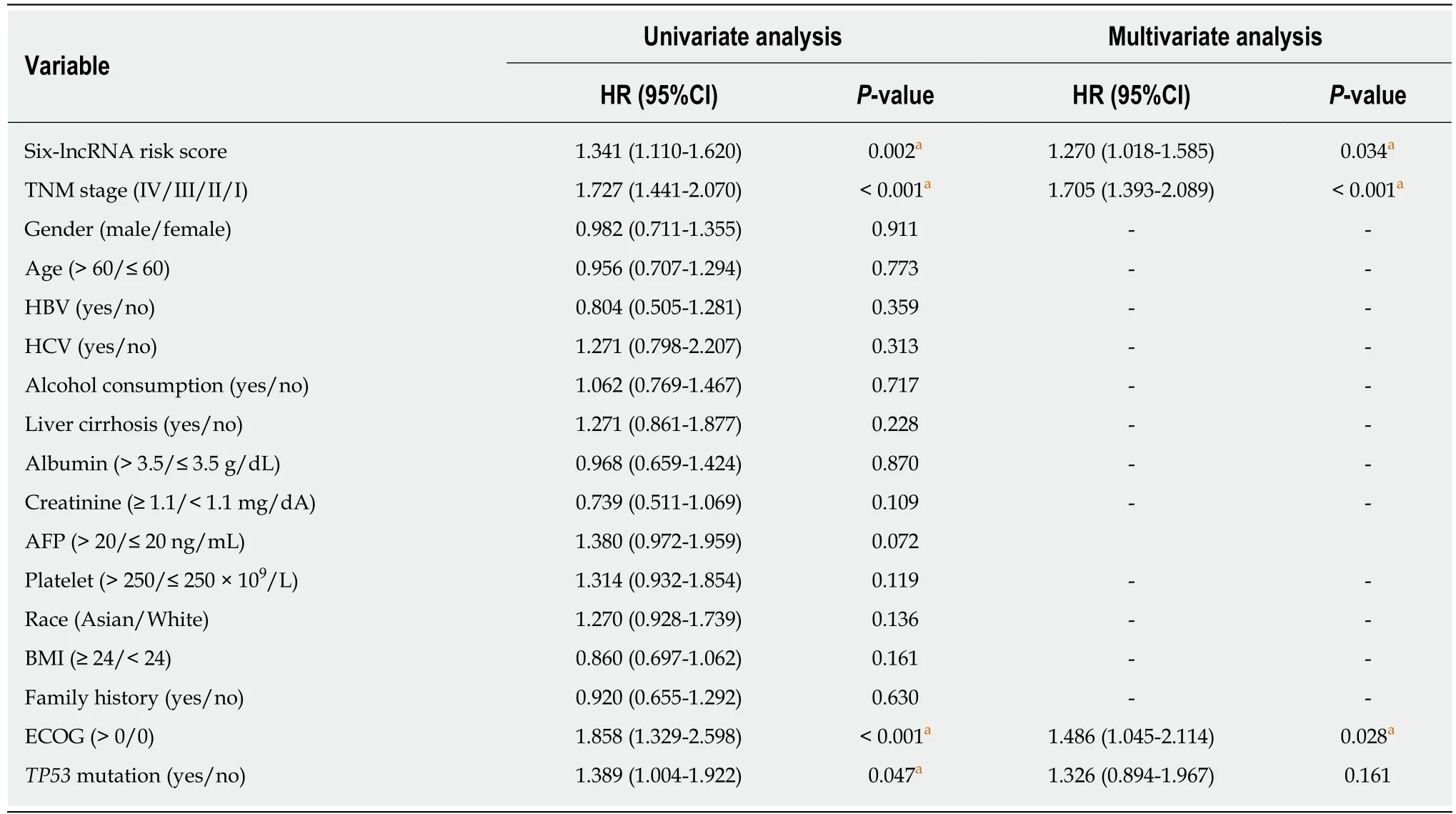
Table 2 Univariate and multivariate Cox regression analyses of clinicopathologic characteristics associated with recurrence in The Cancer Genome Atlas samples
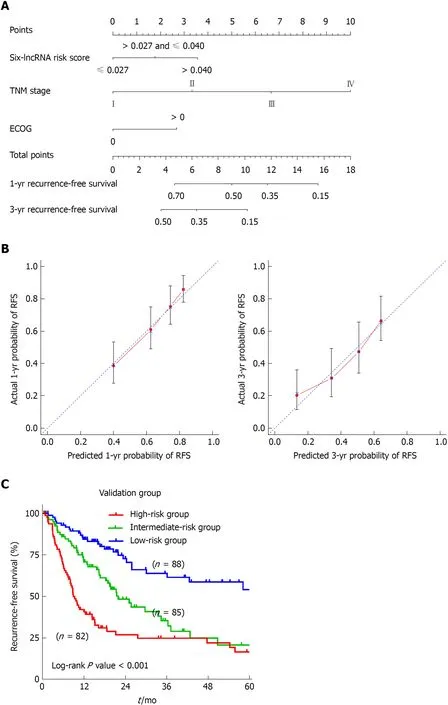
Figure 5 Construction of a nomogram for recurrence-free survival prediction in hepatocellular carcinoma. A: The composite nomogram consists of the sixlncRNA score, TNM stage, and ECOG score. Each component generates their respective points according to the “Points” line drawn above. Add the points from three variables together and find the location of the total points on “Total Points” line. Then draw a vertical line from “Total Points” axis to the two lower lines which corresponds to the predicted 1-year and 3-yr recurrence-free survival (RFS) rates by the nomogram. B: Calibration curves of the nomogram for the estimation of RFS rates at 1-year (Left) and 3-years (Right). The predicted and actual 1-yr and 3-yr RFS-probabilities were drawn on the x and y axis, respectively. C: The Kaplan-Meier curve of three risk subgroups stratified by the tertiles of total points derived from the nomogram.
ARTICLE HIGHLIGHTS
Research background
Hepatocellular carcinoma (HCC) is the most common type of liver cancer which remains a severe health issue worldwide. In recent years, genetic markers and predictive models have been put forward for improving the management of HCC. Meanwhile, many statistical techniques have been used for data mining in a series of large public databases involving the highthroughput genetic data of cancers. With the help of the most advanced clinic-practical methods,more accurate and robust prognostic models can be constructed for HCC.
Research motivation
Researchers have tried to constitute a prognostic model based on molecular biomarkers for HCC over these years. Long non-coding RNAs (lncRNAs) are novel predictive indicators. Although a few attempts have been made to construct lncRNA-based models for HCC, more are needed for further really significant findings.
Research objectives
By analyzing data from two databases, Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA), we wanted to identify a prognostic signature for HCC which is comprised of the potential functional lncRNAs.
Research methods
The latest statistical algorithm, the least absolute shrinkage and selection operator (LASSO), was utilized to constitute our predictive model. This method was performed based on the significant lncRNAs screened based on the lncRNA expression profiles from the GEO database. The expression values of the candidate lncRNAs were also examined in the HCC and normal liver tissues. The robustness of this model was validated using TCGA dataset. The suitable patients and other clinical applicability of the lncRNA-signature were explored as well.
Research results
The risk score system for predicting the recurrence of HCC was constructed based on the six lncRNAs (MSC-AS1, POLR2J4, EIF3J-AS1, SERHL, RMST, and PVT1) using LASSO. All six lncRNAs were aberrantly expressed in HCC and non-tumor tissue and they were significantly enriched in TGF-β signaling pathway and cellular apoptosis-related pathways. The best candidates we identified were younger early-staged male patients with HBV infection and family history in better physical condition but with higher preoperative AFP. To broaden the application scope of the model, a nomogram involving the lncRNA signature and other clinicopathological characteristics was formulated.
Research conclusions
The six-lncRNA signature showed great predictive ability in prognostic evaluation of HCC patients. This tool may help perform risk stratification and provide more individualized clinical advice for each patient.
Research perspectives
Our study offered extra evidence that lncRNAs are potential functional regulators in HCC progression. Finding effective molecular biomarkers and predictive signatures of HCC prognosis are future direction calling urgently for groundbreaking attempts.
杂志排行

World Journal of Gastroenterology的其它文章
- Role of diet and gut microbiota on colorectal cancer immunomodulation
- Current status, problems, and perspectives of non-alcoholic fatty liver disease research
- Prevention of overuse: A view on upper gastrointestinal endoscopy
- Adverse events related to colonoscopy: Global trends and future challenges
- Human antigen R mediated post-transcriptional regulation of inhibitors of apoptosis proteins in pancreatic cancer
- Biomarker identification and trans-regulatory network analyses in esophageal adenocarcinoma and Barrett's esophagus