LC-MS/MS同时测定茭白中阿维菌素和甲氨基阿维菌素苯甲酸盐残留量的不确定度评估
2019-01-28杨德毅吾建祥刘莉刁银军朱丽燕虞冰马婧妤
杨德毅,吾建祥*,刘莉,刁银军,朱丽燕,虞冰,马婧妤
(1.金华市农产品质量综合监督检测中心,浙江 金华 321007; 2.金华职业技术学院,浙江 金华 321017;3.金华市农业科学研究院,浙江 金华 321007)
阿维菌素(avermectin,ABA)是1976年由日本北里大学学者Takahito Omur等和美国Merck公司首先开发研制的杀虫剂,是由灰色链霉菌发酵产生的一类十六元大环内酯化合物;甲氨基阿维菌素苯甲酸盐(emamectin benzoate,EMA)是美国Merck公司1984年在阿维菌素B1基础上合成的一种高效抗生素杀虫剂。其杀虫活性是阿维菌素的103倍,杀虫谱更广[1]。阿维菌素和甲氨基阿维菌素苯甲酸盐在茭白生产中常用于防治绿飞虱、螟虫、蚜虫等虫害[2-4]。GB 2763—2016中茭白中的阿维菌素和甲氨基阿维菌素苯甲酸盐最大残留限量未作规定,但近年来研究表明,该2种农药若不合理使用,对生态环境和人类健康存在较大风险[5-6]。本研究参照NY/T 761—2008和GB/T 20769—2008前处理方法和检测方法,建立了液相色谱-串联质谱(LC-MS/MS)同时测定茭白中阿维菌素和甲氨基阿维菌素苯甲酸盐残留量的分析方法;根据JJF 1059.1—2012《测量不确定度评定与表示》[7]、CNAS—GL06《化学分析中不确定度的评估指南》[8]中规定的程序与方法和相关文献[9-13],对该方法进行不确定度评估,分析不确定度的主要来源,寻求影响不确定度的最大因素,并通过相应的措施减少不确定度对结果带来的影响,为检测机构质量控制提供科学、准确的依据,为用LC-MS/MS测定其他农药残留的不确定度评估提供参考。
1 材料与方法
1.1 材料
测试材料为茭白,系金华市农业科学研究院基地种植的本地品种茭白。
试剂有乙腈(色谱纯)、甲醇(色谱纯)、乙酸铵(优级纯);甲醇中阿维菌素标准溶液(100 mg·L-1±0.6%)、甲醇中甲氨基阿维菌素甲酸盐标准溶液(100 mg·L-1±0.6%),购于农业部环境保护科研监测所;NH2氨基萃取柱,天津博纳艾杰尔科技有限公司生产。
所用仪器与设备有液相色谱-串联四级杆质谱仪(美国Waters公司),T25均质器(德国IKA公司),S-EVAP氮气吹干仪(美国Organomation公司),固相萃取装置(美国Supelco公司),电子天平(瑞士Mettler Toledo公司),超纯水仪(美国Milli-pore公司),涡旋振荡器(美国Thermo公司),移液器(德国艾本德公司)及实验室常规仪器。
1.2 方法
1.2.1 样品前处理
将茭白样品切碎,充分混匀,采用四分法缩分后,经食品加工器捣碎,装入封口容器中,贴上标签,-20 ℃保存,待测。
准确称取25.0 g样品(精确至0.01 g)于250 mL烧杯中,加入50 mL乙腈和50 mL超纯水,匀质器(转速1 300 r·min-1)充分匀浆2 min后经滤纸过滤至装有10.0 g氯化钠的具塞量筒中,滤液剧烈振荡1 min后,静置20 min,用移液器转移10 mL上清液至小烧杯中,氮吹至近干,待净化[14]。
上述浓缩样品用2.0 mL二氯甲烷+甲醇(95 mL+5 mL)溶解后过NH2萃取柱。该小柱先用4.0 mL二氯甲烷+甲醇(95 mL+5 mL)预淋条件化,当溶剂液面到达柱吸附层表面时,立即倒入样品溶液,用15 mL尖底刻度离心管接收洗脱液,用4 mL二氯甲烷+甲醇(95 mL+5 mL)润洗烧杯后淋洗NH2萃取柱,并重复1次。将盛有淋洗液的刻度离心管置于水浴(50 ℃)氮吹仪上氮吹蒸发至近干,用甲醇在刻度离心管内定容至5.0 mL,旋涡混匀,移入2 mL自动进样器样品瓶中,待测[15]。
1.2.2 混合标准液的配制
分别用1 mL的单标线吸量管各吸取1 mL阿维菌素标准溶液和甲氨基阿维菌素甲酸盐标准溶液于10 mL容量瓶中,用甲醇定容,配制成10 mg·L-1的混合标准溶液。
用1 mL的单标线吸量管和10 mL容量瓶逐级稀释上述混合标准溶液分别得到1 mg·L-1、100 μg·L-1、10 μg·L-1混合标准溶液;吸取5 mL 1 mg·L-1混合标准溶液定容至10 mL容量瓶中,得到500 μg·L-1混合标准溶液;吸取1 mL 500 μg·L-1混合标准溶液定容至25 mL容量瓶中,得到20 μg·L-1混合标准溶液;用1 mL的单标线吸量管和10 mL容量瓶逐级稀释500 μg·L-1混合标准溶液分别得到50 μg·L-1、5 μg·L-1混合标准溶液。
按上述方法,得到质量浓度分别为5、10、20、50、100 μg·L-1的混合标准工作液。
1.2.3 仪器条件
色谱柱:Phenomenex Luna(150 mm×2.0 mm,5 μm);柱温:35 ℃;样品室温度:10 ℃;进样体积:5 μL;流动相A:甲醇,流动相B:0.1%乙酸铵水溶液;流速:0.2 mL·min-1,梯度洗脱条件见表1。
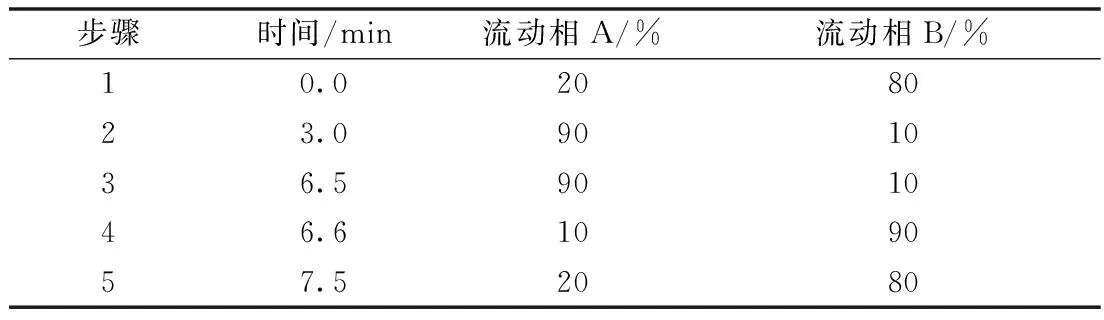
表1 梯度洗脱条件
质谱扫描方式:多反应监测(MRM);离子模式(Ionization Mode):电喷雾正离子源ESI+;毛细管电压(Capillary):3.00 kV;离子源温度(Source Temperature):150 ℃;锥孔反吹气流量(Cone Gas Flow):40 L·h-1;脱溶剂气温度(Desolvation Temperature):500 ℃;脱溶剂气流量(Desolvation Gas Flow):800 L·h-1;母离子m/z:886.2;子离子m/z:158.2(定量离子)、126.1;锥孔电压:80、80 V;碰撞能量:50、62 eV。
1.2.4 数学模型的建立
根据测量的原理建立测量值的数学模型,进行不确定度来源的分析。采用标准曲线法定量,数学模型:
X=cV1V3÷V2÷m。
式中:X为样品中被测组分的含量(μg·kg-1);c为从标准工作曲线得到的试样溶液中被测组分的浓度(μg·L-1);V1为提取溶剂总体积(mL);V2为吸取用于检测的提取溶液体积(mL);V3为试样溶液最终的定容体积(mL);m为试样的质量(g)。
1.2.5 不确定度分量的主要来源
根据该方法测量过程,测定茭白样品中阿维菌素和甲氨基阿维菌素甲酸盐残留量的不确定度的来源主要包括标准品的引入、标准溶液的配制、标准曲线的拟合、样品称量、试样转移和定容、测量重复性、回收率引入等。
2 结果与分析
2.1 单一因素不确定度
2.1.1 标准品引入的不确定度
根据标准物质证书,阿维菌素和甲氨基阿维菌素甲酸盐的纯度误差均为±0.6%;按B类评定取矩形分布,由标准品引入的相对不确定度(urel(sp)):
2.1.2 标准溶液配制过程中引入的不确定度
玻璃量器引入的不确定度。根据JJG 196—2006《常用玻璃量器》[16]检定规程规定的最大允许误差,按以下公式计算标准溶液配制过程中玻璃量具允差引起的相对不确定度(表2),配制过程中所用的玻璃量具均为A级,标准溶液配制过程中引入的不确定度(urel(sv)):
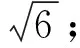

表2 标准溶液配制过程中玻璃量器引入的 不确定度
根据表1中的数据合成标准溶液稀释过程中玻璃量器与移液器引入的相对不确定度为:
urel(sv)=
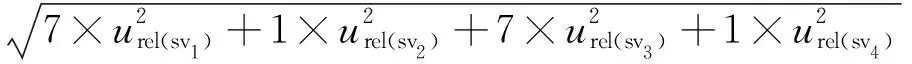
0.011 1。
由环境温度变化引入的不确定度(urel(st))。标准溶液在稀释过程中的溶剂为甲醇,实验温度为(20±5)℃范围内时(温度波动为5 ℃),甲醇的膨胀系数为0.001 19 ℃-1,玻璃量器的膨胀系数远小于液体的膨胀系数,忽略不计,根据以下公式计算环境温度变化引起的相对不确定度,计算结果见表3。
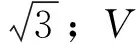
根据表3中的数据合成标准溶液在配制过程中由温度变化引入的相对不确定度:

表3 标准溶液配制过程中温度变化引入的 不确定度
urel(st)=
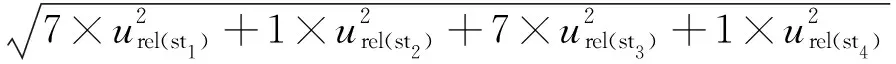
0.013 7。
合成标准溶液配制过程中引入的不确定度(urel(svt))。根据标准溶液配制过程,2 种农药的标准溶液在配制过程中引入的相对不确定度相同,合成 2 种农药标准溶液配制过程由玻璃量具和温度引入的相对不确定度为:
2.1.3 标准曲线拟合引入的相对不确定度
通过对质量浓度分别为5、10、20、50、100 μg·L-1的阿维菌素和甲氨基阿维菌素甲酸盐混合标准溶液测定1次,采用最小二乘法拟合标准溶液的质量浓度与峰面积曲线,各数据结果详见表4。
同时按照上述方法对在田间施用过2种农药的阳性茭白样品进行3次平行测定,测得两种样品溶液平均值c0(ABA)=25.15 μg·L-1、c0(EMA)=108.6 μg·L-1(根据1.2.4节公式计算阳性样品平均残留量X0,结果见表5),根据以下公式对标准曲线拟合引入的标准不确定度进行计算,结果见表4。

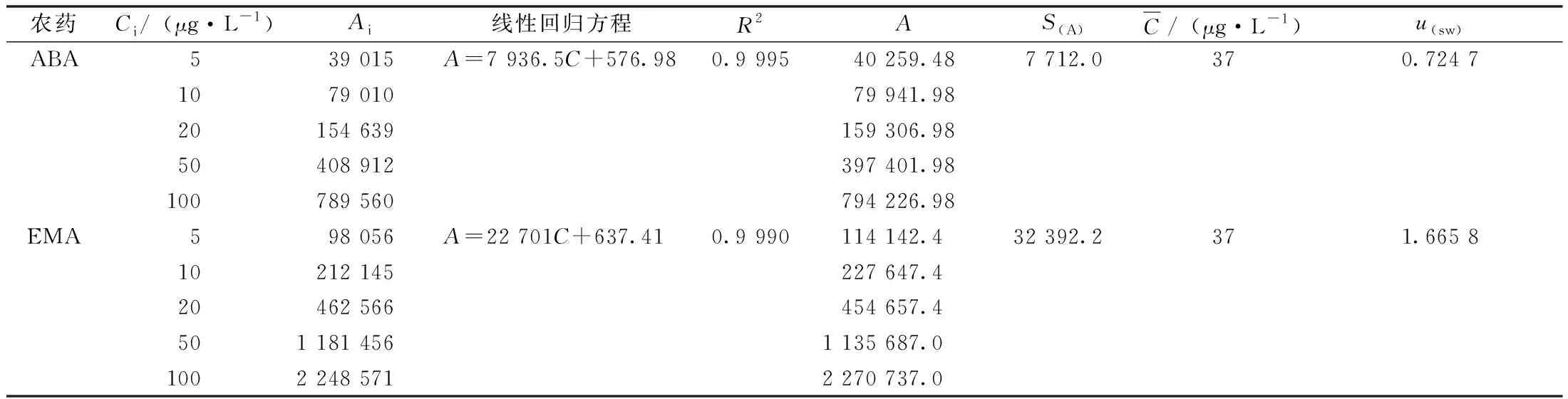
表4 校准曲线拟合数据及不确定度

表5 阳性样品测定结果
2.1.4 试样质量称量引入的不确定度
称量时称取25.0 g(精确到0.1 g)的试样,根据鉴定证书天平最大允许误差为±0.05 g,按B类评定取矩形分布,由试样称量引入的相对不确定度(urel(dm)):
2.1.5 试样转移和定容引入的不确定度
在样品前处理过程中温度变化范围为(20±5)℃,用移液器转移10 mL乙腈溶液(膨胀系数为0.001 37 ℃-1),最终用甲醇在15 mL尖底刻度离心管定容至5 mL,根据JJG 646—2006《移液器检定规程》[17],10 mL移液器的最大允许误差为±0.6%,根据JJG 10—2005《专用玻璃量器检定规程》[18]10 mL尖底刻度离心管最大允差为±0.2 mL,尖底刻度离心管、移液器及温度变化取矩形分布,按上述2.1.2方法分别计算移液器、玻璃量具引入的相对不确定度urel(dv)、温度引入的相对不确定度urel(dt)及合成试样转移和定容引入相对不确定度(urel(dvt)),结果见表6。
由表6数据合成试样转移和定容过程引入的相对不确定度:

表6 前处理过程中移液器和玻璃量具的 相对不确定度
2.1.6 由测量重复性引入的不确定度
在田间施药后取回的阳性茭白样品经3次重复测定的结果见表5,按照农药残留取样及测定的要求,对样品充分混匀、加大样品的制样量及科学缩分方式保障试样的均匀性和检测结果的代表性。同时按A类评定对其不确定度(urel(df))进行计算,计算公式:
计算得出由测量重复性引入的相对标准不确定度为:urel(df(ABA))=0.016 97;urel(df(EMA))=0.003 446。
2.1.7 回收率引入的不确定度
在空白试样中添加 5 组不同的混合标准溶液进行测定,按 A 类进行评定,由回收率引入的标准不确定度(urel(dr))根据如下公式计算,该方法的回收率及标准不确定度结果见表5。
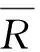
按照以下计算公式对回收率进行显著性检验,对照t检验临界值分布表(f=n-1=4)得双侧临界值t(0.05,4)为2.776。按照以上t值公式计算检验值t(表7)均大于2.776,因此与回收率100%有显著差异,在计算结果时必须采用回收率进行修正。修正后阳性茭白样品中阿维菌素和甲氨基阿维菌素甲酸盐残留量为:

表7 回收率和不确定度
因此由回收率引起的相对不确定度为:
2.1.8 进样体积引入的不确定度
LC-MS/MS 配备的微量注射器体积为 10 μL,相对标准偏差(RSD)为±1%,样品上机进样量为 5 μL,按均匀分布,由体积引入的相对不确定度(urel(ev)):
2.2 复合因素不确定度
2.2.1 合成标准不确定度
LC-MS/MS 法同时测定茭白中阿维菌素和甲氨基阿维菌素苯甲酸盐残留的相对不确定度分量见表8,相对合成不确定度(urel(c))计算公式:
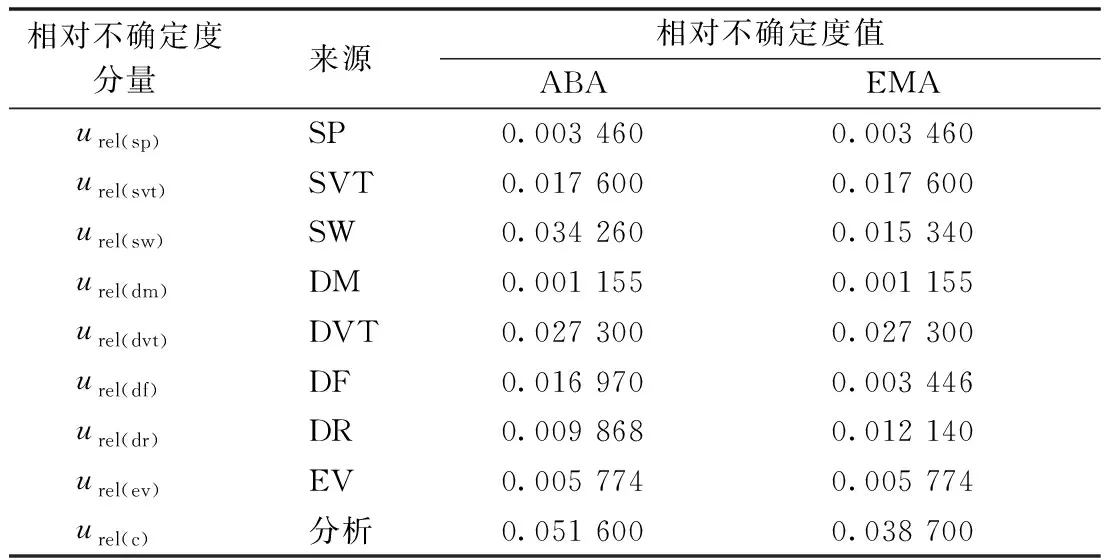
表8 相对不确定度的分量及相对合成不确定度值
注:SP,标准品纯度;SVT,配制标准溶液;SW,标准曲线拟合;DM,样品称量; DVT,试样转移和定容;DF,测量重复性;DR,回收率;EV,进样体积。
分别计算阿维菌素和甲氨基阿维菌素甲酸盐的相对合成不确定度为:
urel(c(ABA))=0.0516 00;urel(c(EMA))=0.038 700。
根据合成标准不确定度的计算公式uc(X)=urel(X)×X0,其中X0为表5中实测阳性茭白样品中残留量的平均值,分别计算得到阿维菌素和甲氨基阿维菌素甲酸盐的合成标准不确定度为:uc(X(ABA))=1.298;uc(X(EMA))=4.203。
2.2.2 扩展不确定度
依据JJF 1135—2005《化学分析测量不确定度评定》[19],在95%的置信水平下,取包含因子k=2,扩展不确定度u=k×uc(X),阳性样品中阿维菌素和甲氨基阿维菌素甲酸盐残留量及不确定度评定结果见表9。

表9 不确定度的评定结果
2.3 影响不确定度因素
从各相对不确定度分量值(表8)看出,用LC-MS/MS法同时测定茭白中阿维菌素和甲氨基阿维菌素苯甲酸盐残留量产生的相对不确定度范围分别在0.001 155~0.034 620和0.001 155~0.027 300,影响相对不确定度主要因素为配制系列标准溶液、标准曲线拟合、试样转移和定容等(图1)。

图1 测定茭白中阿维菌素和甲氨基阿维菌素 苯甲酸盐不确定度来源
3 小结与讨论
通过对茭白中阿维菌素和甲氨基阿维菌素苯甲酸盐残留量测定过程中引入的各相对不确定度评定分析,结果表明,测定茭白中阿维菌素残留量的过程中对不确定度贡献较大的因素依次为标准曲线拟合、试样转移和定容、标准溶液配制;测定茭白中甲氨基阿维菌素苯甲酸盐的过程中对不确定度贡献较大的因素依次为试样转移和定容、标准溶液配制、标准曲线拟合。其中在试样转移和定容过程中相对不确定度较大原因主要为定容过程中刻度离心管最大允许误差较大引入。因此,在实际检测过程中,可通过改进前处理等方法、选择高精密度的量器、提高检测人员业务水平和操作的规范性、选择高纯度的标准品等方法减小测量数据的不确定度,提高检测结果的准确性。
