PU场景下的生物医学命名实体识别算法研究
2019-01-24高冰涛翟振刚
高冰涛 ,翟振刚 ,刘 斌
(1.中国电子科技集团公司第三十六研究所,浙江 嘉兴 314000;2.西北农林科技大学 信息工程学院,陕西 杨凌 712100)
0 引言
近年来,由于生物医学领域命名实体识别研究的潜在应用价值和问题的复杂性,这项研究已经吸引了很多感兴趣的研究者。目前大部分生物医学命名实体的识别主要集中在识别Medline文本中的基因和蛋白质的名称,识别分子生物医学中的命名实体成为生物信息学中知识发现的最基本任务。例如Merry K P和Modi M在文本中提取蛋白质互作信息,第一步就是蛋白质名称的识别[1]。准确高效的生物医学命名实体识别系统对生物医学和生物信息学工作者的研究具有重要的作用和意义[2]。
在传统的生物医学命名实体识别中工作中,由于维特比算法在序列数据中的优良表现和生物医学命名实体的特性,多采用隐马尔可夫模型(Hidden Markov Models,HMM)[3]作为主要算法进行研究和应用[4]。例如:基于单词相似度平滑技术的HMM命名实体识别分类器[5]、PowerBioNE生物命名实体识别系统[6]、BioTrHMM生物医学命名实体识别系统[7],Jie Zhang等人也指出HMM在生物医学领域中进行命名实体识别的有效性[8-9]等。
在生物医学命名实体识别领域中,传统的识别算法为了获得良好的预测性能和保证模型的健壮性,通常要使用大量的标注数据对模型进行训练。但是,在实际应用当中,我们能够直接获得的全标注数据往往很少,并且人工标注数据的成本高昂。PU学习作为一种半监督学习方法,具有比传统的有监督学习方法更大的灵活性。与有监督学习方法相比,半监督学习方法需要的标注数据样本数量少,降低了分类模型对目标领域标注样本的需求量,克服了模型学习过程中由于目标领域标注数据样本不足造成的局限。半监督学习方法,在标注数据不足的情况下,不仅可以保证算法的性能,还有效地节约了资源。
正例未标注学习[10],即PU学习(Positive and Unlabeled Learning),是一种半监督学习方法。PU学习在疾病基因的识别[11]、与时间有关的数据流问题的处理[12]和构建AUC优化方法[13]等方面应用广泛,并且在不确定数据和风险评估方面都取得了非常好的效果[14-17]。
研究至今,暂没有发现研究者在生物医学命名实体识别领域中通过使用PU学习进行研究的相关内容。本文将PU学习有效地应用到生物医学命名实体识别当中,在少量标注数据和大量未标注数据的情况下构建模型,实现命名实体识别。本文主要是从PU学习中的两步法和隐马尔可夫模型的角度展开研究,将生物医学领域中的命名实体识别问题转化为PU场景下的命名实体识别问题,在PU场景下建立隐马尔可夫模型,对命名实体进行识别。
1 问题定义
给定目标领域数据集为D=P∪U,其中P表示数据集中的正例样本的集合,U表示数据集中未标注数据样本的集合,U中同时含有正例样本数据和负例样本数据。每个数据样本可以表示为〈x,f(x),y〉,其中,x 表示观测样本数据;f(x)表示样本 x 对应的词性状态属性,当样本是未标注样本时f(x)取值为?;y∈{0,1},表示样本是否为蛋白质命名实体,y取值为0时表示该样本不是蛋白质命名实体,y取值为1时表示该样本是蛋白质命名实体。本研究目标是使用少量的标注正例样本,对大量未标注的数据分类出强负例样本RN,最后在强负例样本RN和正例数据样本P的基础上学习构建隐马尔可夫模型f,对目标数据集Dt中的样本x有:
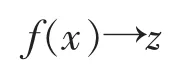
即得到样本对应的词性状态,从而得到特征的取值,进而实现命名实体识别。
2 算法构建
本文将通过使用PU学习两步法构建模型:在使用少量标注数据样本的情况下,通过PU学习构建模型,对待测样本进行预测;通过算法分类未标注数据中的强负例,使用强负例样本和正例样本构建PU场景下的隐马尔可夫模型。技术路线可分为4个主要步骤:数据集收集与预处理、分类出强负例构建分类模型训练数据集、构建分类模型和预测与评估,算法模块的关系如图1所示。
2.1 分类强负例
本文主要使用了Rocchio算法、朴素贝叶斯算法、Spy算法和1-DNF算法等4种方法对未标注数据中的强负例样本进行分类。
(1)Rocchio 算法
在Rocchio分类算法中,每一个文本d用一个特征向量表示,=(q1,q2,…,qn)。 其中,向量中的每一个元素q1表示一个单词wi。Rocchio算法构建模型是通过构建算法中的正例标准向量和负例标准向量实现的,正例标准向量和负例标准向量的计算方式如下:
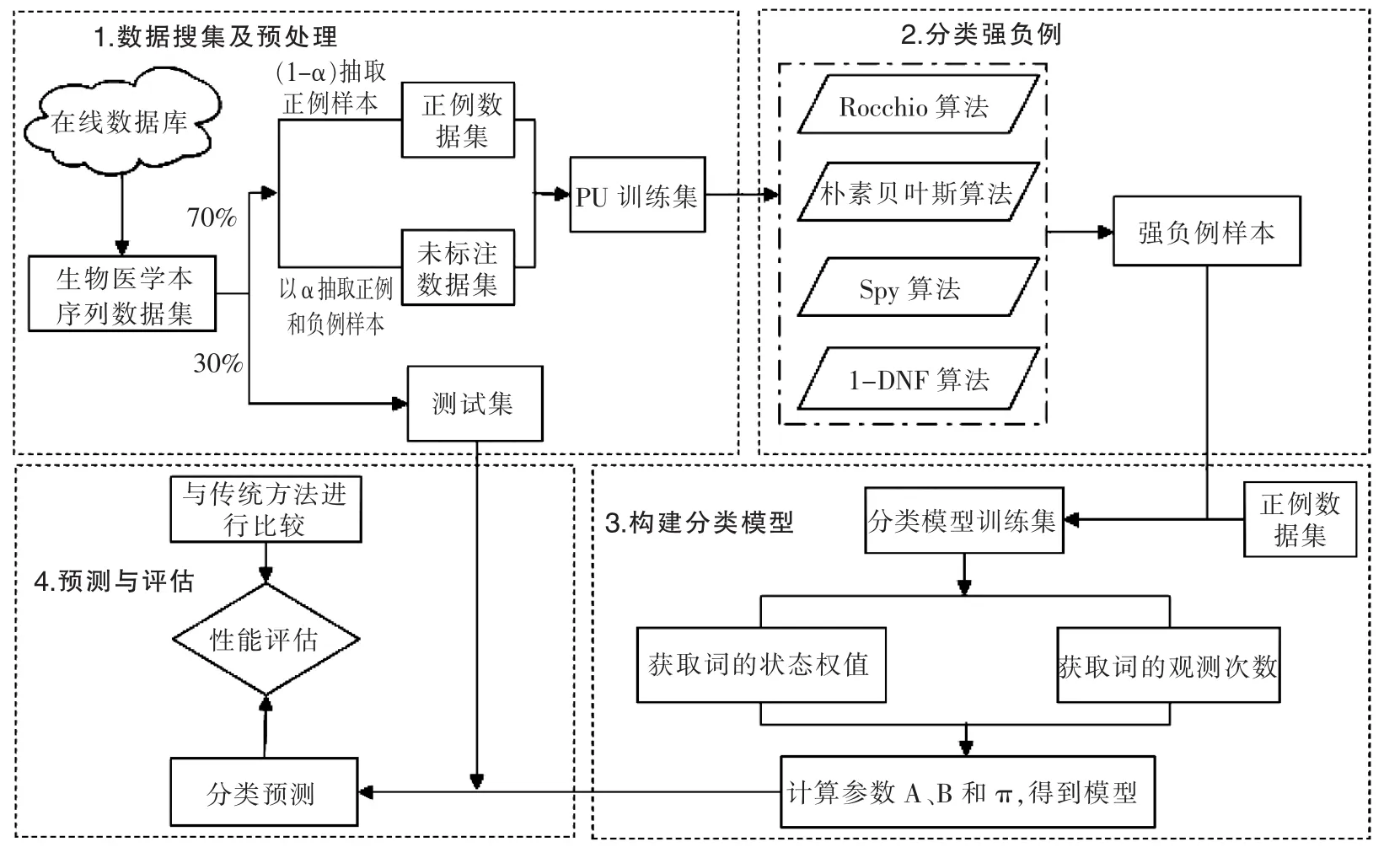
图1 算法模块关系图
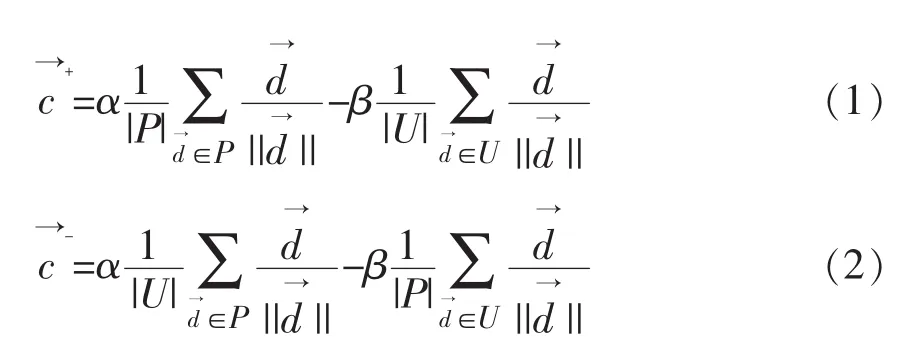
其中,参数α和β是用来对正例训练样本和负例训练样本的相关影响进行调整的,推荐使用α=16,β=4[18]。
Rocchio算法具体如下:

(2)朴素贝叶斯算法
朴素贝叶斯算法在分类问题当中应用非常普遍。对于想要分类的文本集D中的样本数据,C=(c1,c2,…,cn)是预先定义的文本序列数据类别,V=(x1,x2,…,x|v|)表示词汇表,其中 xi表示一个单词。朴素贝叶斯(NB)分类器对给定的文本数据计算条件概率,计算得到的最大概率的那个类别被认为是文本数据的类别。
N(xt,di)表示单词 xt在文本摘要 di中出现的次数,在给定一个类别cj的情况下通过公式(3)计算单词xt出现的概率P(xt|cj),本文中所需要的先验概率P(cj)通过统计得到。
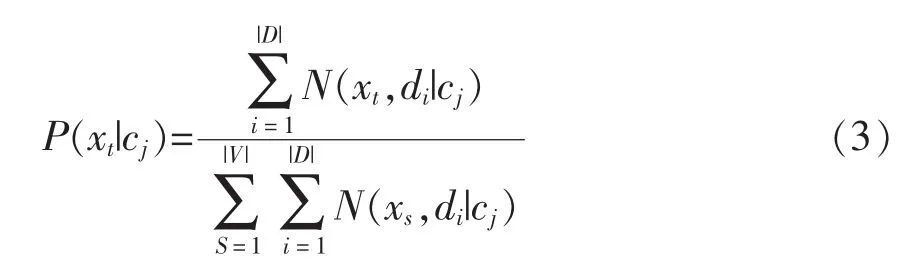
为了防止词汇表V中的某些单词在某些类别的文本当中没有出现,使用拉普拉斯平滑技术进行处理,如公式(4)所示:
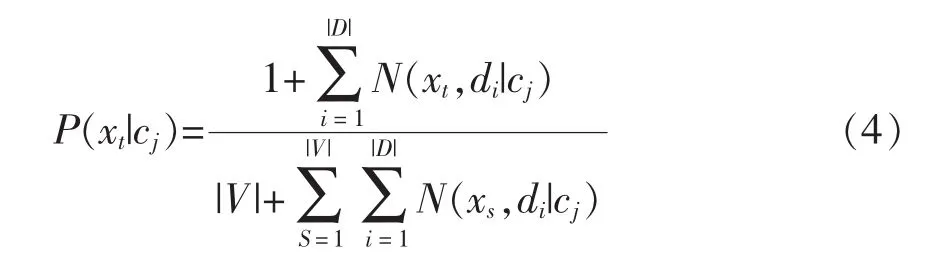
最后,假设给定类别的文本中单词出现的概率相互独立,本文使用式(5)所示的朴素贝叶斯分类器:
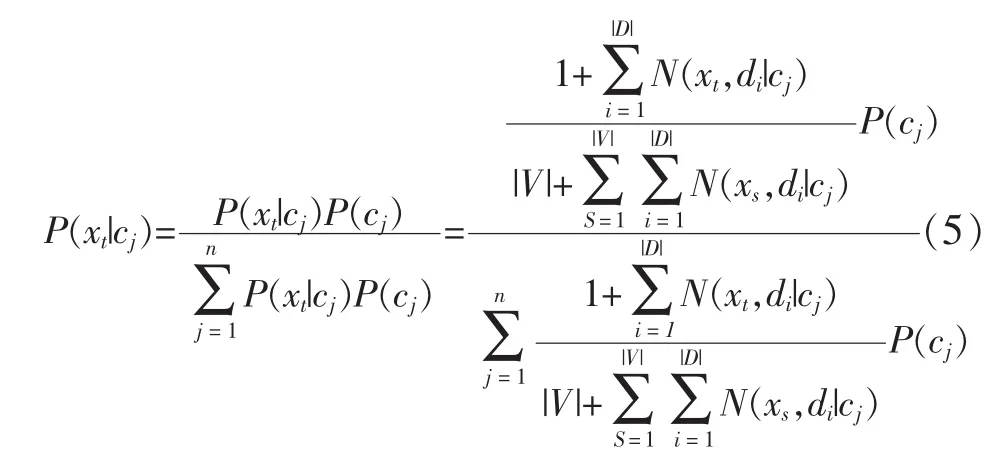
对于给定的文本序列di,通过上述公式计算条件概率P(xt|cj),概率最大的类别作为单词xt的类别。为了从未标注数据U中识别出强负例数据集RN,本文在正例数据集P和未标注数据集U上训练一个朴素贝叶斯分类器,并且用该分类器对未标注数据U进行分类。对于给定的文本数据,如果是正例的概率小于是未标注数据的概率,则把文本数据看做是一个强负例样本。
朴素贝叶斯分类器的算法框架如下:

(3) Spy 算法
Spy算法见算法3。对在Spy算法中使用的概率阈值t进行简单说明:计算“Spy”数据S中每一个样本分布为正例的概率,取其中的概率最小值作为概率阈值t。
Spy算法的具体过程如下:

(4)1-DNF 算法
1-DNF算法是通过对正例样本数据P和未标注样本数据U中的数据特征进行对比,找到一些正例样本所具有的的特征,构建一个正例特征集PF。该方法计算在正例样本数据P和未标注样本数据U中单词出现的频率,然后使用在正例样本数据P中出现频率比在未标注样本数据U中出现频率高的单词构建正例特征集PF。该方法是通过对未标注数据U中的所有数据样本进行核查,将其中可能是正例数据的样本筛选出来,这样未标注数据U中的不含任何正例特征的样本就被分类为强负例样本。
1-DNF算法的具体过程如下:

2.2 构建分类模型
通过上述4种方法可以有效地从未标注数据中将强负例样本分类出来,进而与正例样本形成训练集,这样就可以进行有监督学习。由于HMM在命名实体识别研究中的有效性,本文同样选择HMM作为基础模型。HMM的三个主要参数分别是初始状态概率向量π、状态转移概率矩阵A和观测概率矩阵B。
转移概率aij的估计。设文本序列样本中时刻t处于状态i,在时刻t+1转移到状态j的频数为Na,ij,则状态转移概率 aij的计算公式如下:

观测概率bj(k)的估计。设样本中状态为j并且观测为k的频数为Nb,jk,则状态为j观测为k的概率 bj(k)的计算公式如下:

初始状态概率πi的估计。i为S个样本中初始状态为qi的频率,计算公式如下:

由于可能出现的数据稀疏问题,本文使用拉普拉斯平滑的方法进行处理,具体如下:
初始状态概率计算为:
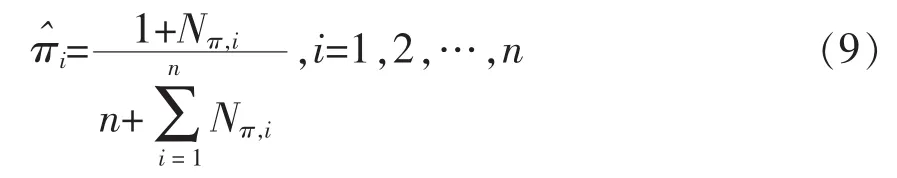
转移概率aij的计算为:
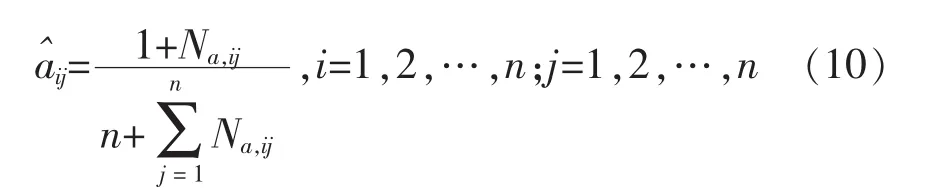
观测概率 bj(k)的计算为:
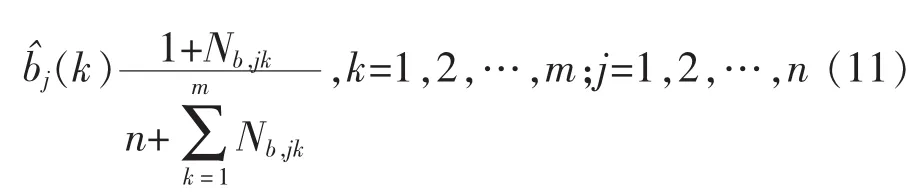
至此得到分类预测模型,并使用维特比算法对待测数据进行标注,从而对命名实体进行识别。
3 实验及结果分析
为了验证本文提出算法的性能,本文在GENIA V3.02语料库上进行了实验。
3.1 实验设置
为了验证本文提出的算法在生物医学领域中命名实体识别的性能,选取传统的HMM算法与本文提出的PU场景下的两步法算法进行比较。目前,最常用的生物医学标注语料库是GENIA V3.02语料库,该语料库包含了来自MEDLINE的2000个摘要标注文本(约360000个单词),并且包含36个词性类别,其中包含5个生物医学实体类型。本文识别的是蛋白质命名实体,采用了精确率、召回率和F值[19]作为评价指标。GENIA V3.02语料库中实体标签分布说明见表1。

表1 GENIA V3.02语料库中实体标签分布
本文中Dt是含有蛋白质命名实体标签和其他词性标签的目标集,Ds是把蛋白质命名实体标签处理为NN类型的辅助集,辅助集中标签分布见表2。

表2 辅助集中实体标签分布
本文采用PU学习中一种普遍使用的方法[20]构造PU数据集。对一个数据集,正例样本以概率(1-α)随机选择标记为正例,这部分样本构成正例样本集,剩下的正例样本作为未标注样本,这部分样本和所有的负例样本构成未标注样本集。
根据He J[21]等提出的实验方法,为了测试在不同PU场景下算法的预测性能,本文设置了两个参数α和Unlevel来模拟不同的PU场景。α表示正例样本占源数据集的比例;Unlevel表示未标注样本占源数据集的比例。

本文通过对每组实验进行十折交叉验证的方法,确保结果的有效性。
3.2 实验结果
为了验证PU场景下两步法算法的性能,本文分别从不同角度进行了实验。实验如下:
(1)针对 α的实验
本文分别设置参数α为0.2,0.4,0.6,对PU情况下通过两步法训练构建的HMM与直接使用现有的少量标注数据构建的HMM的分类性能进行了对比,实验结果如表3至表5所示。
实验结果显示,标注数据较少的情况下,使用两步法构建的分类模型比直接使用现有标注数据构建的分类模型性能更优。当参数α为0.2,0.4和0.6时,通过PU学习方法构建的分类模型比直接使用现有的少量标注数据构建的模型在准确率和召回率方面具有显著的优势。同时,通过PU学习得到的分类模型的准确率和召回率虽然随着参数的变化有所起伏,但是总体变化不大,比直接使用现有的少量标注数据构建的分类模型更加稳定。
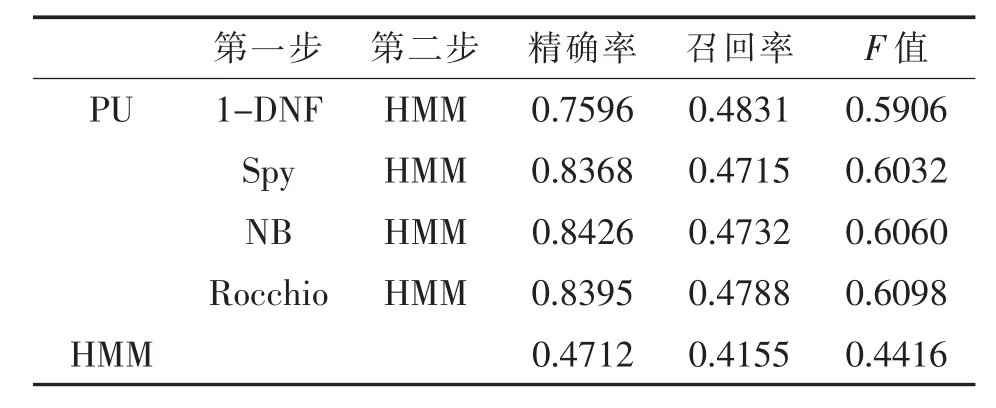
表3 α=0.2时分类性能对比
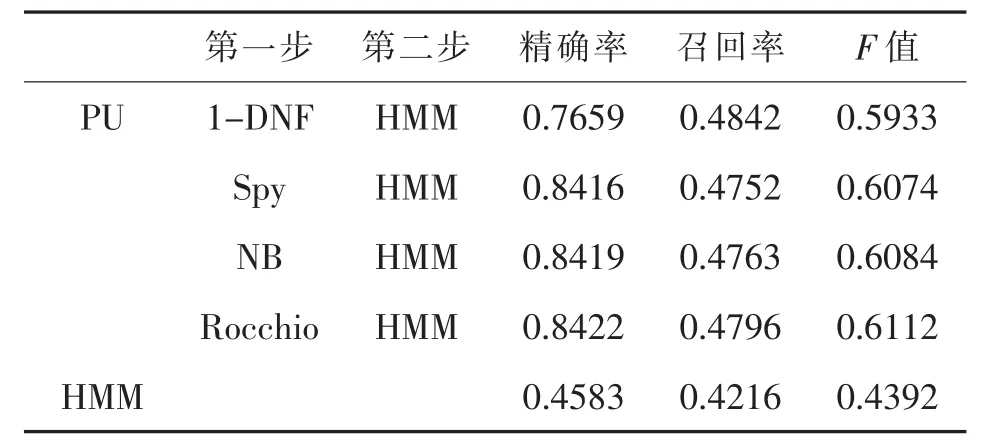
表4 α=0.4时分类性能对比
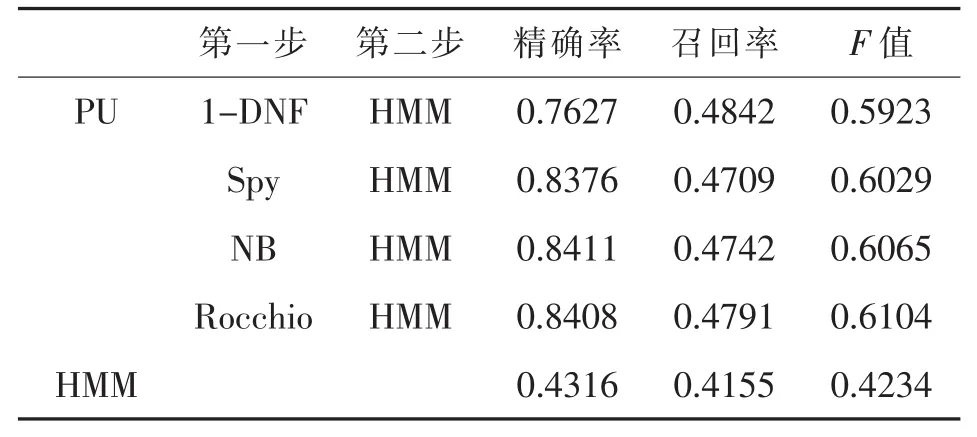
表5 α=0.6时分类性能对比
(2)针对 Unlevel的实验
本文将Unlevel分别设置为50%,60%,70%,80%,90%。对在PU情况下通过两步法训练构建的HMM与直接使用现有少量标注数据构建的HMM的分类性能进行了对比。表6至表10是模型在不同的Unlevel情况下的实验结果对比。
本文在不同的PU学习情况下,分别使用1-DNF、Spy、NB和 Rocchio算法作为两步法的第一步,然后在第二步中使用已有正例数据和分类出的强负例数据训练HMM。实验结果显示,在标注数据较少的情况下,本文通过两步法得到的模型比直接使用已有标注数据训练得到的分类模型具有更好的分类性能。并且在模型实验结果的准确率和召回率方面,与直接学习得到的分类模型相比优势明显,特别是在未标注样本比例逐渐增大的情况下,优势越加显著。
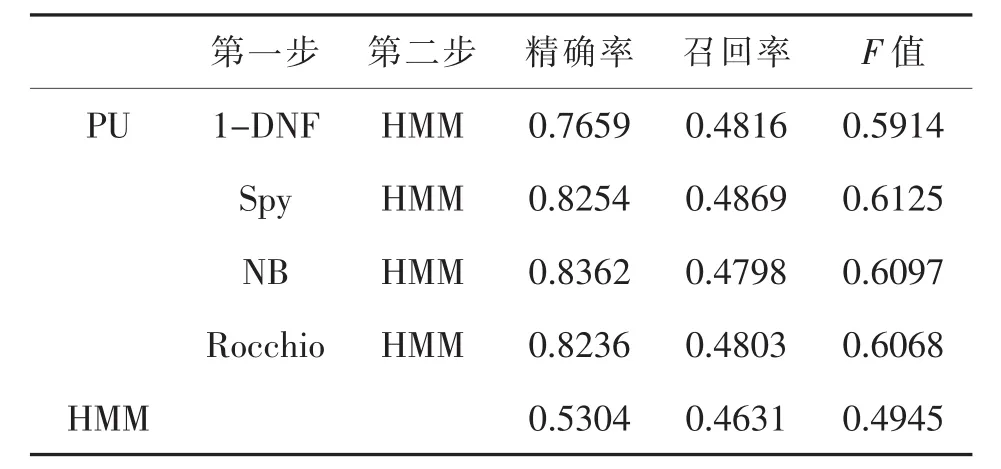
表6 Unlevel=50%时分类性能对比
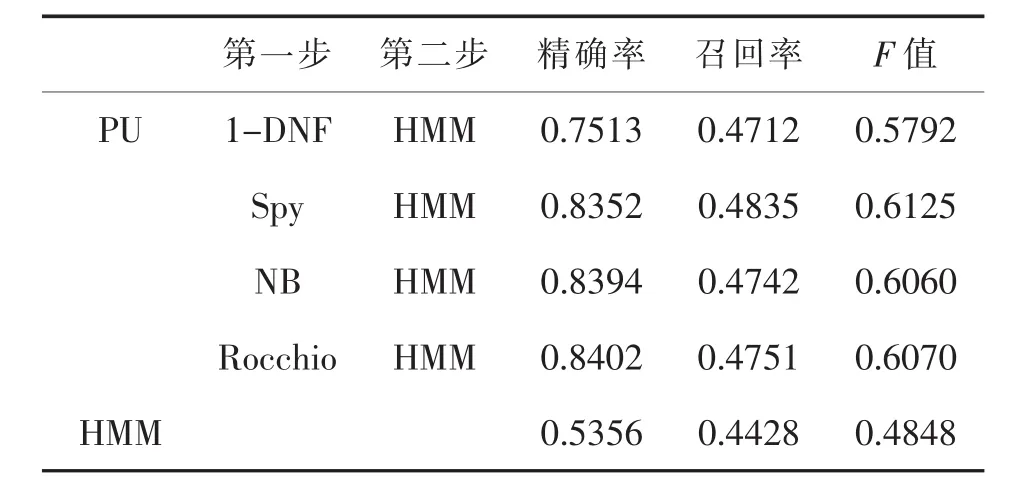
表7 Unlevel=60%时分类性能对比
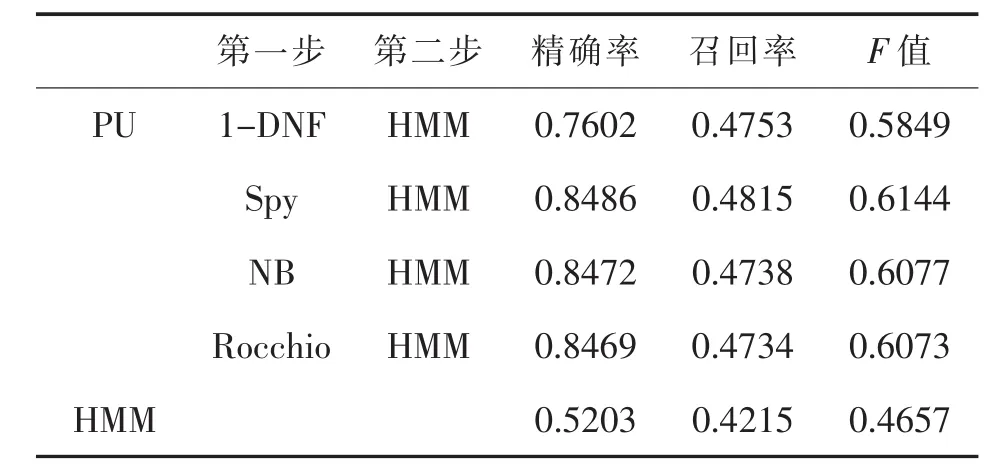
表8 Unlevel=70%时分类性能对比
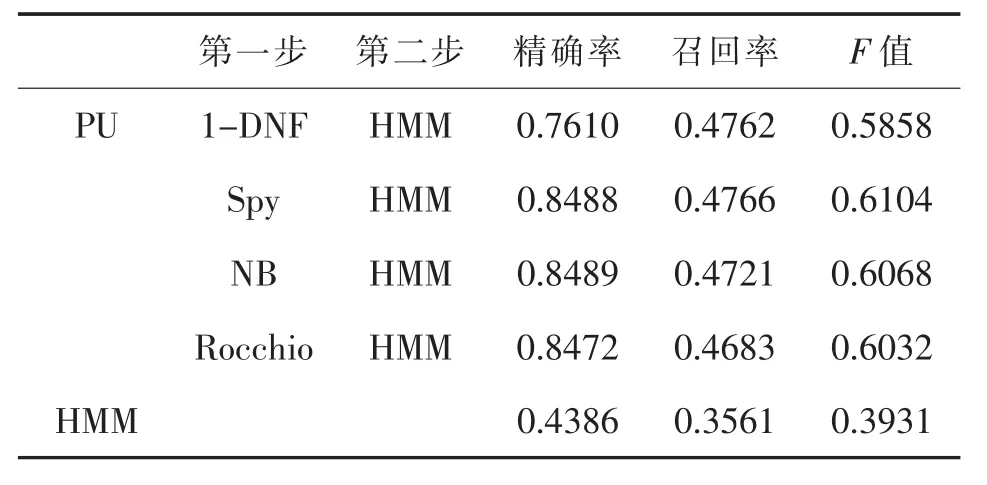
表9 Unlevel=80%时分类性能对比
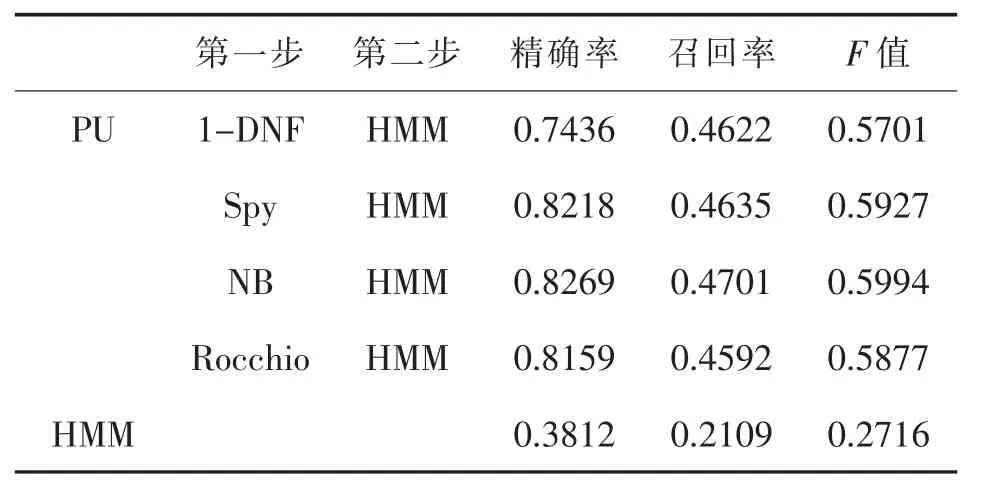
表10 Unlevel=90%时分类性能对比
通过设置不同的参数α和Unlevel值,模拟不同PU场景下的生物医学领域的蛋白质命名实体识别问题。以上的实验结果表明,在不额外增加人工标注目标数据的情况下,通过两步法构建的分类模型比直接使用现有的少量标注数据构建的分类模型具有更好的分类性能。
4 结语
针对传统生物医学命名实体识别方法需要大量标注数据,而人工标注数据困难、能获取标注数据比较少的问题,本文提出PU情况下通过两步法构建分类模型的生物医学命名实体识别方法。通过PU学习方法中的两步法在未标注数据中分类出强负例样本,在已有的正例样本和分类出的强负例样本的基础上对模型进行训练,构建出分类模型,对目标数据进行命名实体识别。实验显示,在只有少量标注数据的情况下,通过PU学习中的两步法构建的分类模型比直接使用现有少量标注数据的监督学习方法构建分类模型具有显著优势。此外,通过PU学习方法构建分类模型不仅识别性能有所提升,同时大大节省了人工标注数据的成本。
在本文中主要通过PU学习方法构建分类模型,降低模型对标注数据的需求。现在深度学习方法的研究越来越热,逐渐涉及到多个领域,未来工作考虑在生物医学命名实体领域对深度学习方法进行研究。
