不同阶段阿尔茨海默病患者海马CA1区基因表达的生物信息学分析*
2019-01-18宋祯彦余婧萍贺春香陈易璇李富周成绍武
宋祯彦,余婧萍,贺春香,陈易璇,李富周,成绍武
(湖南中医药大学中西医结合心脑疾病防治湖南省重点实验室 长沙 410208)
1 前言
阿尔茨海默病(Alzheimer's disease,AD)是与衰老相关的致死性中枢神经系统退行性疾病。根据阿尔茨海默病协会2016年发布的1份报告,每33秒就会出现1 例AD 新病例,每年新增病例近100 万例,预计到2050 年,该病的流行范围将达到1100 万到1600 万[1]。AD 是由复杂的病因引起神经功能障碍,其发病的分子机制尚不明确[2]。目前大多数人认为AD 的发病过程与脑内高密度的老年斑和神经纤维缠结有关,但目前针对上述发病环节的药物并不能减缓或阻止AD 的进展[1]。因此,需要进一步研究AD 的发病机制以制定有效的治疗策略。
据报道大约70%的AD 发病危险因素是由遗传引起的,通常涉及多个基因[3]。随着分子生物学技术的发展,基因芯片能同时对数千个基因的表达进行分析,可有效地用于研究与AD 发病相关的基因网络[4]。不同阶段的AD 患者、同一阶段AD 患者的不同脑区,其基因表达谱有很大差异[5,6]。大脑海马区主要负责记忆和学习,AD 中海马是首先受到损伤的区域,表现症状为记忆力衰退以及方向知觉的丧失,经缺血缺氧后,海马CA1 区发现明显的神经元损伤而出现空间定位和学习记忆功能障碍[7]。基因表达综合数据库(Gene Expression Omnibus datasets,GEO)(https://www.ncbi.nlm.nih.gov/gds)中的基因芯片数据GSE28146 包含从早期到晚期AD 的一系列与认知功能障碍和神经纤维缠结症状相对应的海马CA1 区基因表达的变化[8]。我们据此来分析AD 不同时期大脑CA1 区细胞的基因表达变化规律,期望找到在AD 不同时期共性表达的关键基因,以进一步阐明AD 发病机制并为寻找新的治疗靶点提供依据。
2 材料与方法
2.1 细胞
SH-SY5Y 细胞株购自中国科学院上海生命科学研究所。
2.2 主要试剂
β-淀粉样蛋白1-42(amyloid β-protein 1-42,Aβ1-42,A9810)购自上海荣创生物技术有限公司;噻唑蓝(Thiazolyl Blue Tetrazolium Bromide,MTT,ST316)购自碧云天生物技术有限公司;二甲基亚砜(Dimethyl sulfoxide,DMSO,D806645)购自上海麦克林生化科技有限公司;TRIzol(15596-026)购自美国Invitrogen 公司,反转录cDNA 试剂盒(PrimeScript™RT reagent Kit with gDNA Eraser,RR047A)购自日本TaKaRa 公司;SYBR 染料(SYBR Premix Ex Taq II,RR820L)购自日本TaKaRa公司。
2.3 主要方法
2.3.1 GEO数据挖掘
从基因表达综合数据库GEO 中下载编号为GSE28146 的芯片数据原始文件,该芯片数据来源于早期、中度、重度阿尔茨海默病患者,包含总样本数30 例,其中正常8 例,早期7 例,中度8 例,重度7 例。利用激光显微切割术获取石蜡包埋的大脑海马CA1区灰质组织,基于美国昂飞公司人类基因组U133+2.0 芯片平台(GPL570: [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array)的基因表达谱进行芯片数据采集分析。本研究利用Robust Multiarray Average(RMA)算法对芯片数据进行了背景校正和矩阵数据归一化处理,同时采用limma 包对芯片数据进行二次分析,并结合P值和差异倍数(fold change,FC)进行显著差异基因筛选,筛选条件为P<0.05,FC >2。
2.3.2 AD 患者不同时期(早期、中度、重度)差异基因的获取
使用R 语言分析包以P<0.05,FC >2为筛选条件分别筛选AD 早期患者与正常对照、AD 中度患者与正常对照、AD 重度患者与正常对照的显著差异基因。合并筛选AD 3 个时期的差异基因,获取在AD 早期、中度、重度均出现显著差异的重合基因,利用Venny2.1(http://bioinfogp.cnb.csic.es/tools/venny/)构 建韦恩图。
2.3.3 蛋白互作网络(protein-protein interaction network,PPI network)构建和关键基因筛选
PPI network 可用于过滤和评估功能基因组数据,并对理解功能基因的产物蛋白的相互作用及其影响的主要生物学功能提供一个可视化的平台[9]。探索预测AD 不同时期共同的DEGs(differentially expressed genes)的PPI 网络可以为寻找AD 发生的重要作用靶点以及未来的防治方案提供新的方向。本文利用相互作用基因/蛋白质检索在线数据库STRING(https://string-db.org/)[10]来寻找AD 不同时期共同的DEGs 的相互作用(score ≥0.4),然后使用Cytoscape-v3.6.1 进行构建网络图。在此基础上,对PPI 网络进行拓扑学特征分析,通过Cytoscape 的插件CytoNCA 计算参数来评价网络中每个节点在功能上的重要性,筛选参数分别为中间中心性(Betweenness centrality,BC)和接近中心性(Closeness centrality,CC),度中心性(Degree centrality,DC)、特征向量中心性(Eigenvector centrality,EC)、基于局部平均连通性的方法(Local average connectivitybased method,LAC)、网络中心性(Network centrality,NC),这6 个典型的中心属性参数用以进行评价网络节点的重要性,值越大说明节点越接近网络中心位置,即关键基因[11]。
2.3.4 关键基因的Gene Ontology和pathway富集分析
基因本体论分析(Gene Ontology,GO)(包括生物过程(BP)、细胞组分(CC)、分子功能(MF))和KEGG通路富集分析是描述候选基因生物学特征的常用方法[12]。GOBP 是生命活动的重要体现,GOBP 富集分析可以体现疾病发生发展的重要生物学过程[13]。我们利用(DAVID,https://david.nicifcrf.gov/)数据库进行功能注释和富集分析,以P-Value <0.5 为参数获得关键基因的GOBP富集条目。本研究利用生物信息学在线分析平台Omicshare(http://www.omicshare.com)以P<0.5、FDR <0.05 为参数获取关键基因的pathway 富集信息。
2.3.5 细胞造模及细胞活力检测
SH-SY5Y 用含10%胎牛血清,100 U·mL-1青霉素和100 U·mL-1链霉素的DMEM/F12 培养基在37℃,5%CO2的条件下培养。当细胞生长处于对数期时,按8×103个/孔的细胞密度转移到96 孔板中,培养12 h 使其贴壁,分别设立正常组、Aβ1-42造模组(1.25,2.5,5,10,20,40 μmol·L-1)。各组细胞培养24h 后加入5 mg·mL-1MTT 溶液10 μL,37℃孵育4 h 后弃上清并加入DMSO 200 μL 每孔,酶标仪上震荡30 min,490 nm 波长处测定吸光度A,细胞的生存率=(A 实验组-A 调零孔)/(A空白组-A调零孔)×100%,每组均设置3个复孔。
2.3.6 关键基因的表达qPCR验证
细胞培养方法如1.5,按5 × 105个/孔的细胞密度转移到6孔板中培养12 h使其贴壁,分别设立正常组、Aβ1-42(10 μmol·L-1)造模组,培养24 h 后提取总RNA。总RNA 提取使用TRIzol, 反转录获取cDNA 使用
TaKaRa PrimeScript™RT reagent Kit with gDNA Eraser。采用SYBR Premix Ex Taq II 和CFX96 荧光定量PCR仪(Bio-Rad,美国),按照试剂盒说明书对筛选出的5个关键基因进行qPCR 验证。所用引物序列从NCBI数据库获取,使用Primer Premier6.0 软件设计并由华大基因有限公司进行合成(表1)。
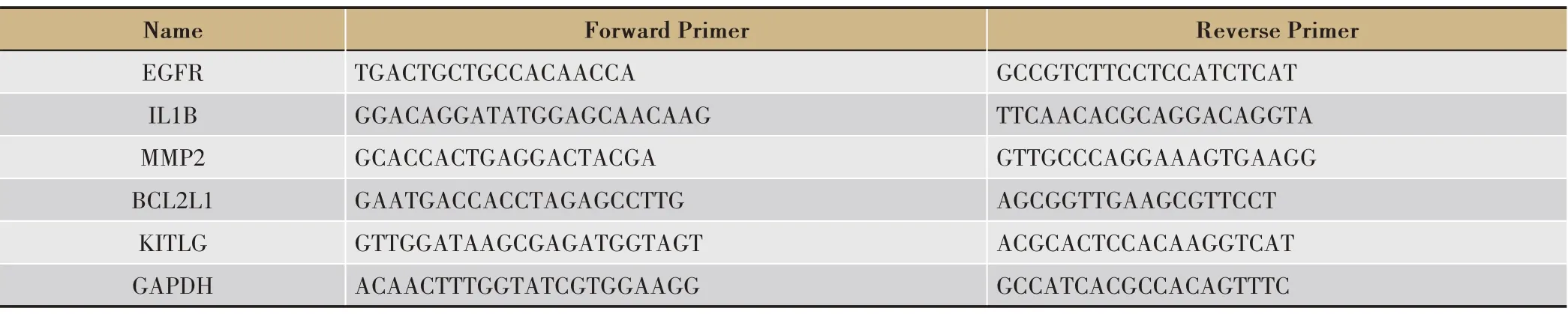
表1 引物序列
2.4 统计方法
采用Prism GraphPad 6.0 统计分析软件进行数据统计分析,计量资料以±s表示,采用t检验,P<0.05为差异有统计学意义。
3 结果
3.1 AD 患者不同时期(早期、中度、重度)差异基因的分析
二次挖掘分析GEO 芯片数据库的基因芯片GSE28146,以P<0.05,差异倍数(Fold change,FC)>2 为筛选分析AD 早期患者与正常对照组的基因差异表达情况,芯片结果显示共有2314 差异表达基因;AD中度患者与正常对照组比较共有1782 个基因差异表达;AD 重度患者与正常对照组比较存在929个显著差异基因。其中,在AD 三个时期均存在差异表达的基因有419 个(如图1),其中上调基因有235 个,下调基因有184个。
3.2 AD差异基因的PPI网络分析及关键基因筛选
将419 个与AD 相关性蛋白靶点导入到STRING数据库获取这些靶点的PPI 网络(图2),其中249 个蛋白之间存在498种相互作用关系。采用CytoNCA 计算PPI 网络的BC、CC、DC、EC、LAC 和NC 值并进行筛选(图3),这些参数中DC 值大于两倍中位数值的节点为重要节点,所以第一次筛选采用DC 大于两倍中位数即DC >4 进行筛选,得到初步筛选出的网络,该网络有86 个节点,285 个边,在此基础上以DC >8.3489、EC > 0.0675、BC > 172.8372、CC > 0.3414、NC >3.8023、LAC >2.5763 对网络进行第二次筛选,得到核心网络[11,14]。该网络有12 个节点存在48 种相互作用关系,因此该12个核心靶点确定为AD发生过程中的关键基因(表2)。
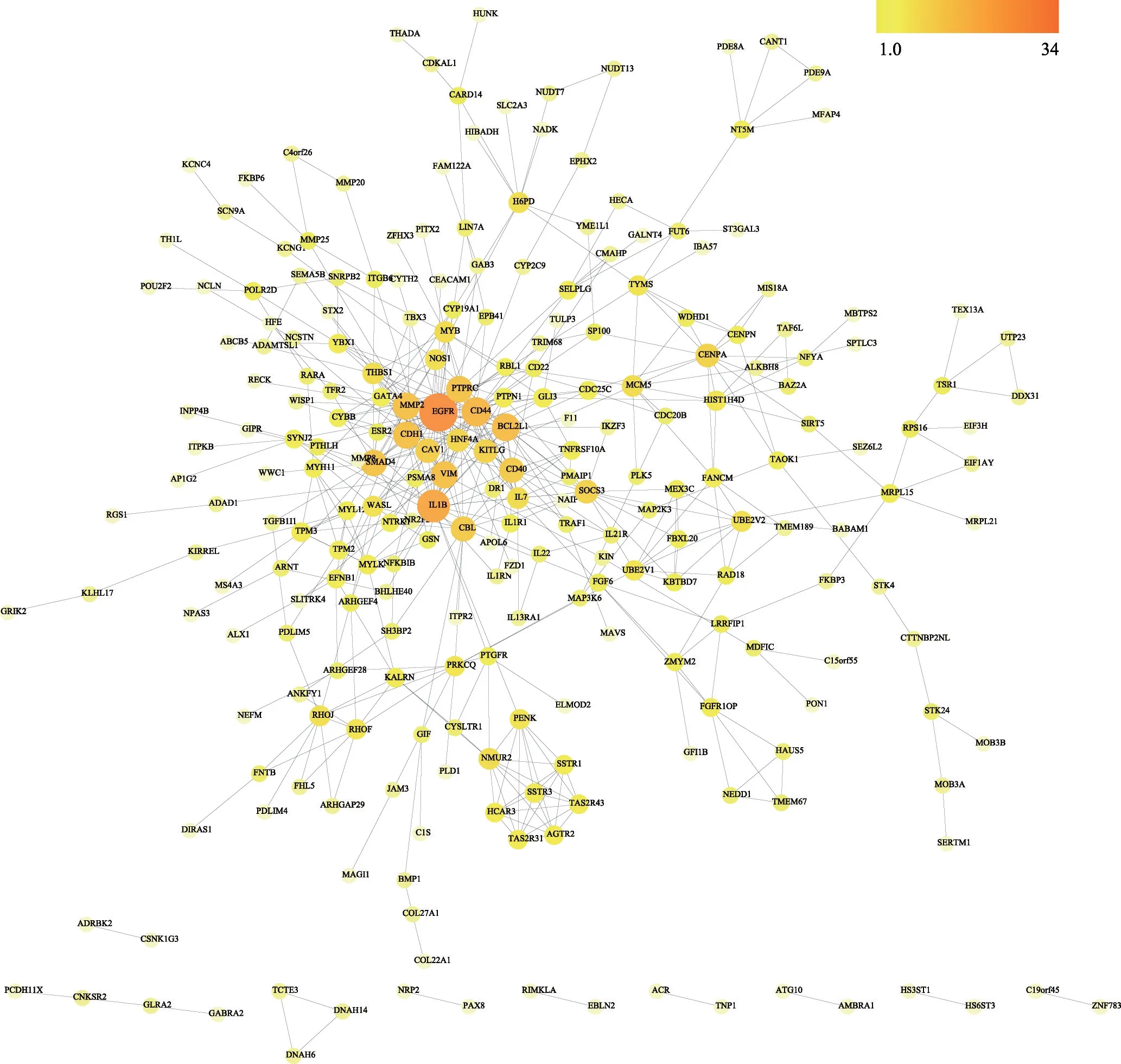
图2 AD相关的蛋白靶点的PPI网络图

图3 关键基因的PPI网络分析.

表2 关键基因的差异表达(AD VS. Control)

图4 关键基因的富集分析
3.3 关键基因的GO分析和pathway分析
为了进一步了解关键基因的作用,通过GO 和KEGG 通路富集分析来初步揭示它们在AD 发生过程中起到的生物学作用。结果表明靶点参与的生物过程为一氧化氮合酶活性调节、细胞凋亡、缺氧反应、细胞间黏着、聚集过程、蛋白质乙酰化、MAPK 复合物调控、细胞因子介导的信号通路调控以及胰岛素受体信号通路调控(图4A)等,其中一氧化氮合酶活性调节、细胞凋亡、MAPK复合物调控等都为AD发生过程中的关键过程[15]。此外,这些关键基因主要参与了Rap1 信号通路、Ras 信号通路、NF-κB 信号通路、TNF 信号通路、PI3K-Akt 信号通路、Phospholipase D 信号通路、Jak-STAT 信号通路、Calcium 信号通路(图4B)。根据文献报道,在这些通路中,Ras 信号通路和PI3K-Akt信号通路在AD 发生过程中起着重要作用,AD 发生过程中Ras表达增加激活糖原合成酶激酶3(GSK-3),导致APP和Tau过度磷酸化,引起AD 大脑中的β淀粉样蛋白沉积和神经纤维缠结[16,17]。同时Ras 蛋白作为信号通路的分子开关,调控下游PI3K-Akt 信号通路和MAPK信号通路,参与氧化应激、自噬、凋亡、炎症反应等多种生物学过程[18,19]。而PI3K-Akt 信号通路、NFκB 信号通路、TNF 信号通路等参与神经炎症反应,与AD发生也密切相关。
3.4 AD细胞模型构建及关键基因的qPCR验证
SH-SY5Y 细胞衍生于人的神经母细胞瘤细胞系SK2N 2SH,该细胞分化后具有神经元样的形态,表达神经元特异性的标志物,表现出人神经元的特性,该细胞系被广泛运用于神经退行性疾病发病机制的研究。本研究以不同浓度Aβ1-42刺激SH-SY5Y细胞构建AD 细胞模型,MTT 检测细胞活力显示在Aβ1-42刺激后SH-SY5Y 细胞活力随浓度呈下降趋势(图5A),Aβ1-42浓度为10 μm·L-1刺激下细胞生存率为55 ± 6.8%,故选择10 μm·L-1Aβ1-42建立AD 细胞模型。TRIzol 法提取细胞总RNA 逆转录cDNA 后,使用设计好的引物进行qPCR 验证。结果显示,与正常组比较,Aβ1-42处理后EGFR、MMP2 表达明显下降(P<0.05),IL1B、BCL2L1、KITLG表达显著上调(P<0.05)(图5B),挑选的5个关键基因qPCR表达结果与芯片结果一致。

图5 AD细胞模型的建立及关键基因的qPCR验证
4 讨论
大脑海马区属于脑的边缘系统(limbic system)中的重要结构,与学习、记忆、认知功能有关,尤其是短期记忆与空间记忆[20]。大量研究证实海马的损伤会导致长期情景记忆的严重损害,这表明海马在学习和记忆中起着重要作用[21,22],特别是CA1 区被认为是长时程增强效应形成的关键部位,因为它是介于海马和皮质/皮质下区域之间的信息通道[23,24]。AD 患者与非痴呆患者的大脑相比,海马CA1 区通常表现出明显的萎缩,神经元和突触的数量减少[25]。近年来,有学者提出CA1 区的病理改变是认知障碍患者记忆功能障碍的重要指标[26]。
目前普遍认为,在AD的病理过程中,tau蛋白过度磷酸化导致的细胞内神经原纤维缠结(neurofibrillary tangles,NFTs)、细胞外淀粉样蛋白(β-amyloid protein,Aβ)积聚所形成的老年斑是AD 发生的主要病理生理机制[27]。为了进一步了解核心基因在AD中的作用,我们从GEO数据库获取了不同时期AD患者大脑CA1区灰质的基因表达谱,运用生物信息学方法筛选出在AD 发生的不同时期均出现表达异常的基因并构建了PPI 网络,这些基因的异常表达引起蛋白相互作用网络调控异常可能是导致AD 出现病理特征的关键所在[28],因此我们进一步分析PPI 网络获取了网络中相互作用最多的12个关键靶点,这些关键基因的失调可能是导致AD 发生的重要因素,它们也可能是治疗AD的潜在生物标志物或药物靶点。
为了进一步明确关键基因在AD 发生过程中发挥的生物学功能,我们利用DAVID 数据库和Omicshare在线分析工具对这些关键基因进行生物学过程和代谢通路的富集分析,发现它们主要与一氧化氮合酶活性调节、细胞凋亡、缺氧反应、炎症反应等密切相关。我们结合文献检索分析结果预测AD 的不同阶段海马CA1 区差异表达的基因与AD 发生的联系主要为:①神经炎症反应及相关通路的调控是影响AD 发生发展的重要机制。Aβ 的沉积和神经纤维缠结引起神经元炎症反应,神经炎症反应的激活伴随着许多促炎因子,如肿瘤坏死因子α(TNF-α)、白介素1-β(IL-1β)、白介素6(IL-6)以及活性氮及活性氧的释放,促炎因子毒性作用的累积会导致慢性神经炎症,从而引起神经元死亡[29]。炎性细胞因子的释放又可以反过来促进β 淀粉样前体蛋白的表达,进一步促进AD 的发生[30]。②PI3K-Akt 信号通路、MAPK 信号通路、以及NF-κB信号通路在内的炎症反应途径介导了神经炎症的发生机制。多种介质和信号通路之间复杂的相互作用产生各种炎症因子和神经炎性反应促进AD 的发生发展。细胞外介质可调节不同的信号级联,导致神经元细胞生存或死亡,发挥最终的神经系统功能,其中PI3K/AKT、MAPK 在激活NF-κB 中发挥重要作用[31,32]。在AD 进展过程中,NF-κB 作为转录调节剂在炎症,神经元存活,分化,细胞凋亡,神经突生长,突触可塑性发挥重要作用[33]。在AD 中,Aβ 刺激促使NF-κB 被激活,产生大量的促炎细胞因子,进而作用于神经元和星形胶质细胞,诱导其表达更多的细胞因子和趋化因子,形成恶性循环,也间接促进了神经元死亡[34]。
本研究采用生物信息学方法分析不同阶段AD 患者海马CA1 区差异表达的基因,以阐明AD 发生的分子机制。结果表明一些基因的表达失调可能是导致AD 发生的重要因素,这些关键基因可能通过神经炎症影响AD 的发生发展,证实了神经炎症促进AD 的发病的关系,对AD 治疗的潜在生物标志物或药物靶点的研究有重要意义。
