基于de Bruijn图的基因组索引结构设计
2019-01-11国宏哲王亚东
国宏哲, 王亚东
(哈尔滨工业大学 计算机科学与技术学院, 哈尔滨 150001)
0 引 言
HTS技术的一项重要挑战是将测序序列快速且准确地比对到一个或多个参考基因组上。随着HTS技术的普及而产生的数据量越来越大,序列比对是HTS分析中计算量最大的步骤之一。read比对过程关于速度的要求正日渐提升。而且随着越来越多的新基因组的陆续面世,更新的HTS数据分析需要将reads比对到多个基因组、而不是单基因组上。一个物种的典型链的基因组可以作为金标准,再利用来源于宏基因组样本的reads比对到这些参考基因组去识别物种类型和病原体类型[1-3]。同时,de Bruijn图是代表和组织基因组序列的基本数据结构,并已广泛应用于各类序列分析的学术领域中,如de novo基因组装、高通量测序(NGS)序列比对、泛基因组分析、宏基因组学分类、转录本同种型鉴定和定量分析、NGS序列校正等。这些也是许多基因组学研究的基础。
时下,多数位居前沿的先进比对软件都是根据单基因组进行设计并优化的。同时也有很多方法被提出索引pan-genome[4-5],且都是通过共线性多序列比对技术组织基因组。大部分索引pan-genome的方法只能用于小规模的变异研究,例如软件GenomeMapper[4]和BWBBLE[5]只能整合SNP和短Indel变异。同时,基于共线性结构的比对速度也较慢,其速度比目前较快的比对软件慢几十倍[5-6]。该方法的适用范围仅限于高度相似的多基因组,但还不能针对来源于不同链或不同物种的多基因组发挥效用。这些非同源性的基因组中经常包含结构变异且共线性结构不能提供诸如大型结构变异的非线性序列变换。共线性结构本身也影响序列比对结果,不适合将序列比对到多个不同基因组。产生式压缩后缀数组索引[6]会随着变异数量增加而呈指数增长,使其很难处理在重复区域的变异信息[7]。增广多基因图结构(population reference graph, PRG)[8]可以更好支持不同种类的变异。该方法是将所有reads构建成de Bruijn图和PRG进行比对。其本质是将基因组序列比对和从头拼接相结合。该方法主要是为局部区域而设计适合解决基因组上复杂区域上的相关问题并提高了基因组复杂区域的比对质量。但目前尚未见到有效的方法索引全基因组的PRG结构,还不能将此方法延伸至全基因组上的序列比对。为解决以上方法的限制并优化处理基因组上大量重复片段引起的问题,本文提出一个基于de Bruijn图的索引结构DBG-index来有效组织重复序列,从而实现单基因组和多基因组的索引和序列比对计算。
1 基于de Bruijn图的索引结构分析
1.1 基于哈希表结构的数据存储方法
De Bruijn图结构可以利用hash table数据结构进行存储,即将各个k-mer节点提取指定bp数的2-bit位表示(A/C/G/T分别编码成二进制00/01/10/11)作为地址空间中的地址值。该地址记录k-mer对应入边信息和出边信息,及指向其对应的位置信息的地址,如图1所示。利用de Bruijn图构建索引并实现序列比对的优势如下。某一k-mer序列在基因组中可能出现在多个位置上,de Bruijn图中某一节点表示的k-mer只出现一次且该节点对应的所有位置信息记录在一个数据结构中。在基于后缀树结构和基于BWT结构构建的索引中,每个后缀在基因组中对应唯一的一个位置,其位置信息是分散的,但de Bruijn图中节点将位置信息集合在一起且允许有序。可以进行相邻节点对应位置集合的合并操作以快速筛选提取真实位置信息,并验证当前路径是否正确。通过对现在映射系统原理的分析,发现基于seed-and-extension思想方法[9-11]比对过程中对候选位置进行的extension局部比对是相对耗费时间的计算步骤。如何有效地缩小候选位置的范围、减少extension的次数对系统速度实现至关重要。而基于de Bruijn图结构的遍历过程会大大减少候选extension的位置集合数量。
de Bruijn图结构的一个特点是可以实现双向图,即某一个节点既可以记录其出边,也可以记录其入边(基因组上k-mer序列对应的上一个bp)。该特性可以使种子的延伸方法更加灵活,可以采用双向延伸的方法,从而更快速获得较长的种子和真实的候选位置。
Fig.1Anexampleofgenomick-mersstorageinthehashtablestructure
1.2 基于哈希结构的序列比对方法
k-mer长度越长,在比对过程中需要搜索k-mer的次数越少,且速度也越快。但在实际应用中需要严格控制k-mer的长度,因为地址空间是随着其序列长度的增加而呈指数增长。而且,k-mer长度过大会使比对时seed过长而较大概率地跨过mismatch或indel存在的bp位置,将真实的seed漏掉而无法得到真实的位置信息。如果k-mer长度过小会使整个de Bruijn图过密、节点过多,且每个节点对应的入边和出边也较多,这会使图上的搜索过慢。故k-mer长度的选取成为一个关键问题。针对此问题,需要计算不同k-mer长度在不同物种基因组上的分布。
很多物种的基因组数据的k-mer长度变化趋势,开始快速增长到一个值后趋于稳定。其中,hg18人类基因组数据快速增长到20后,开始缓慢增长,一直到40后趋于稳定。
结合Hash实现过程中的实际情况,针对人类基因组数据使用,本文将序列映射系统索引和比对过程中k-mer长度范围定为21~28,将前14 bp作为地址,并将剩余bp构建连续的数组结构(地址空间约228* 4 B = 1 GB)。
基于Hash索引结构进行序列比对的基本原理如图2所示。从read上从头向后提取k-mer序列转换成地址编码,在Hash索引中进行搜索获得对应的获选位置,在对应位置提取序列和read做局部或全局比对。这也是基于seed-and-extension的经典流程。
基于de Bruijn图的比对过程与此有所不同,在获得某节点的位置信息后会有合并过程,通过此有序位置数据间的合并过程可筛去大量无意义的位置信息,如图3所示。某一位置集合里的位置数值加上read上偏移,在相邻节点对应位置集合中搜索找到合理的位置数值,从而实现两集合间的合并,按照此方法依次向下合并。在合并过程中考虑到mismatch或indel存在的可能性,根据测序序列的错误率和各个类型变异的发生概率设定一个误差范围。
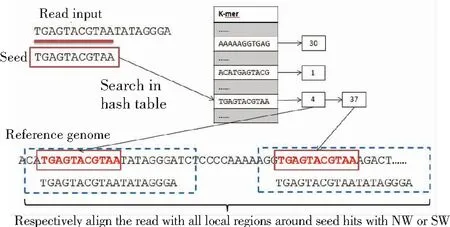
图2 基于hash结构存储的read比对过程
Fig.2Anillustrationofthereadalignmentbasedonthehashstructure

图3 种子对应位置集合合并过程
Fig.3Anillustrationoftheprocessofseedsmergingbythepositioncollections
1.3 k-mer的3层存储结构分析
各部分索引结构设计思想如图4所示。其中包含一个3层结构:第一层是hash的地址空间保存k-mer的前14 bp序列信息;第二层记录后面剩余的k-mer信息,空余出来的信息位可用于记录该k-mer是否有反转操作等信息(用于双向seed延伸操作使用)及入边和出边信息;第三层是k-mer对应的有序位置信息数组。
为实现该索引结构,系统需要遍历一遍基因组序列。且第二层序列内容存储和第三层位置信息需要有序存储,故需要先将基因组上所有k-mer进行排序。而由于基因组序列较长无法做到将所有k-mer统一提取排序,故采用分段排序的办法、即按照字母表逻辑大小顺序(A 图4 索引存储三层结构 需要注意在临时文件中也记录了k-mer对应的位置偏移信息及其对应的入边和出边信息。然后按顺序重新读取每个临时文件,即某一时刻只有一个临时文件的全部k-mer调取至内存中,对这些k-mer生成排序。在此过程中也可以同时构建索引的3层结构:每当临时文件中k-mer排完序后从头遍历该有序结构,每当遇到一个不同k-mer序列,即将该k-mer添加到Hash地址空间中并添加入边和出边信息;若当前k-mer序列与前一个k-mer序列相同,则在k-mer序列对应的位置信息地址后添加新位置信息。 在比对部分首先导入索引文件。某一k-mer的搜索在第一层地址空间中时间是O(1);在第二层后半部分,k-mer序列搜索过程时间是O(n);第三层返回的位置信息数组是由第二层结构中的指针加以控制。其做到了最快速地返回某一k-mer的所有相关信息并实现了位置信息的融合且有序。基于此结构的read的seeding过程可阐释如下。 (1)从read头入手,提取k-mer作为seed开始在Hash索引结构上根据各节点位置集合做延伸停止后,从停止处的下一个bp开始再延伸。 (2)按此方式一直到read遍历完成,将所有获得的seed对应的位置结合再做进一步合并,检验是否有seed间可以合并的可能。 (3)将最终得到的seed按种子长度逐一排序,依次提取seed对应位置集合中位置在基因组上的序列做局部比对或全局比对;若某2个相邻seed得到的比对分数收敛或设定某一固定的结束条件,则终止局部比对的extension环节输出比对结果。 本文构建一个(Reference de Bruijn Graph)结构去组织一个或多个reference并用到基于hash table的数据索引结构DBG-index去索引de Bruijn图上的所有unipaths,而不是索引原始基因组。DBG-index组织和索引reference示例,一个或多个reference被组织到。一个k-mer的所有拷贝被定位到de Bruijn图上的同一个节点,基因组上序列相同的重复片段被定位到相同的unipath上。该新基因组的索引方法提供2个基本操作去识别和合并相似种子,并且利用de Bruijn图上unipath的性质识别相同的局部序列。此处unipath定义满足如下条件:路径起始节点的入度大于1、且出度是1;路径的结束节点入度是1、且出度大于1;路径中间节点入度是1、且出度是1。索引可以同时处理多个相似种子和相同局部序列,从而有效解决基因组的重复性带来的问题。 比对系统是利用索引相关功能设计推出了在图索引上的基于seed-and-extension思想的序列映射算法。算法很好处理基因组上的重复片段,从而极大减少了序列比对的时间开销。用户可以根据自定义k-mer长度构建reference的de Bruijn图。de Bruijn图上的所有节点和边是通过reference上的所有k+1-mers计算获得。遍历de Bruijn图的hash table结构,若当前k-mer为一个起始节点、即开始根据其出边获得其相邻的下一个k-mer,在hash table结构中定位该k-mer,并按此方法循环直至当前k-mer为结束节点,此时可形成一个unipath序列并开始下一个unipath的生成。按此方式遍历完hash table即可生成所有的unipath。一个unipath在基因组上可能有多个拷贝,故需要记录每一个unipath对应的所有拷贝在基因组上的真实起始位置。索引结构包括以下3个主要结构:线性表记录所有unipath序列且为每一个unipath指定一个标识;一个hash table索引unipath上的所有k-mers;线性表记录每个unipath的所有拷贝的起始位置。而且通过该索引结构可实现给定k-mer获得其所在的unipath和在unipath的偏移位置;给定一个unipath和某偏移位置获得在基因组上原始拷贝位置集合。 根据de Bruijn图的原理,某一个k-mer在图结构上只出现一次,任意一个指定k-mer所在的unipath和坐标是唯一的。且某个k-mer在基因组上的位置集合可通过线性表联合计算获得。利用以上功能接口可以通过实现以下2个主要操作(相同unipath上的相似seeds合并;unipath上相同序列合并)去合并相似种子、以及减少相同的extension计算。 某一k-mer在reference上的所有拷贝位于图结构上的同一节点,该性质表明一个k-bp长的seed在reference上的映射等同于连接seed和图结构上的特定节点。seed的命中是一段短片段精确比对到一个unipath的多个拷贝上。通过以上数据结构可计算某拷贝在unipath上的起始位置。 某一read映射到基因组上的某一可能位置可以被估算成某unipath拷贝在基因组上位置加上对应seed在unipath上的位置偏移。多个seeds在同一个unipath则可能有2个相似的候选位置集合且可以将相似位置集合予以合并。可以利用线性表判断任意2个seed是否映射到同一个unipath,如果是同一unipath,可以通过发现seeds间的相似性从而进行相似seeds的合并而不需要分别计算每一个命中位置。在此基础上,考察某一seed在其unipath上的偏移和到unipath的2个端点的距离,从而判断该unipath序列是否容纳read序列进行局部比对。如果可以,unipath对应的所有拷贝和read的所有extension计算就都将归结为read和unipath序列本身的局部比对,而不需要对seed的每一个命中分别进行extension计算。 为测试基于索引结构的seeding(种子获取过程)效果,研究分别统计模拟数据和真实测序数据上的seeding中各过程产生的seeds数,并使用BWA比对软件中的基于BWT索引结构的索引和基于BWT MEM索引结构(maximal exact matches,MEM)的索引进行比较分析。实验中拟使用Mason模拟器分别模拟生成基于Illumina平台的长度为100 bp 和 250 bp的reads的数据集(Sim-i100和Sim-i250)。 模拟数据集上的索引结构seeding统计结果和测序数据集上的索引结构seeding 统计结果可分别详见表1和表2。由表1可见,索引结构对应的seeding过程中第一部分表示初始产生的seeds个数;第二部分表示相同unipath上seeds合并后的seeds数;第三部分表示unipath上的相同序列合并后的seeds数。BWT MEM索引对应的seeding过程中第一部分表示初始产生的seeds数;第二部分表示经过MEM计算后的seeds数。Filtering过程表示各索引结构自身定义的一些seeds的过滤规则,例如,索引中将最长seed长度小于当前read长度一半的seeds过滤掉。不同的过滤规则筛选出的seeds会有所不同。模拟数据集上Valuedseedsratio定义为产生正确比对位置的seeds占经过filtering后的seeds总数的比例;测序数据集上Valuedseedsratio定义为产生合理比对位置的seeds占经过filtering后的seeds总数的比例。 表1模拟数据集上的索引结构seeding统计结果分析 Tab.1StatisticalanalysisofDBG-indexbasedseedingonsimulationdataset DatasetMethodSeeding #/×106Filtering #/ ×106 Valued seeds ratio/%Sim-i100DBG-index4.904.604.504.1048.50BWT-index178.90178.901.10BWT MEM-index32.6019.2012.5015.90Sim-i250DBG-index11.309.908.905.7034.50BWT-index871.70871.700.23BWT MEM-index68.4044.308.1024.80 表2 测序数据集上的索引结构seeding统计结果 由表1和表2可看出,各索引结构随着各个计算过程seeds逐渐减少。模拟数据集和真实数据集上A索引在各个过程生成的seeds最少且最后的Valuedseedsratio最高。BWT索引在各个过程生成的seeds最多且最后的Valuedseedsratio最低。因为MEM方法有种子长度扩展和筛选功能,BWT MEM索引相对BWT索引生成更少seeds。A索引的seeding的有效性是BWT索引的几十倍。相对于BWT MEM索引生成的seeds数目,A索引同样表现出优势,且在Sim-i100上效果优势更加明显。因为read长度较短时可用的seeds数也相对较少,由此即说明当seeds数较少时,索引能更快速地生成正确seeds。这对于后续能够高效实现序列映射算法应用将十分重要。 研究使用来源于千人基因组计划中的样本NA12878的Illumina测序平台生成的ERR174324真实数据集测试各索引结构seeding情况。由于真实数据中的测序错误,也加入了更多、更复杂变异,从表2可看出,整体上真实数据比模拟数据上产生更多的seeds。所有的Valuedseedsratio相对模拟数据降低,同样由于测序错误等问题使索引生成很多错误seeds。在该情况下,索引仍然表现出较好的seeding结果。 在seeding过程中每个seed都有其对应的命中(hits)数,表示在基因组上对应的位置个数。由于基因组上有大量重复片段,一个seed通常对应多个hits。在映射算法中考虑到速度的要求,需要对hit设定一个阈值以实现整个系统在准确度或敏感度和速度上的平衡。为此,研究将在测序数据集上测试不同hits数对seeding情况的影响。研究得出人类基因组上的相关统计结果见表3。其中,Hits-n表示hits数上限阈值是n。随着n的增大,seeds数增大且Valuedseedsratio降低。可以观察到在Hits-300~-700范围内,seeds数增长平稳,且在Hits数大于1 000时seeds数趋于平稳。此类统计分析有利于辅助映射算法选择默认hits阈值。 表3测序数据集上索引结构的不同hits数量阈值下seeding统计结果 Tab.3StatisticsofDBG-indexbasedseedingwithvarioushitsvaluethresholdsonsequencingdataset statisticsHits-100Hits-300Hits-500Hits-700Hits-900Hits-1 100Seeds #/×1071.161.271.311.341.371.37Valued seeds ratio/%17.2015.7015.2014.9014.5014.50 本文首先阐述基于hash table结构的一般基因组索引结构和基于此结构的比对方法。接下来分析了不同长度的k-mer在基因组上的分布情况。然后探讨了seed-and-extension的基本思路,并发现根据位置集合对种子进行合并可以有效减少extension的计算量。 本文首次提出基于hash思想的用于k-mer存储的3层索引结构,并以此为基础提出基于de Bruijn图的索引结构来组织基因组中的重复序列。 同时,提出该索引的基本构建方法和数据的存储格式。之后提出的结构上的PRP集合合并操作、相同的unipath上的种子合并方法和unipath上的相同序列合并方法,表明索引可以极大减少用于extension过程计算的候选种子的数量。 本文分别在模拟数据集和真实数据集上测试索引结构和基于BWT的索引结构的seeding效果。通过比较分析seeding过程中各步骤中的seeds数量情况,发现de Bruijn图结构能减少更多的候选seeds,同时保持更多的有效seeds比率。基于de Bruijn图思想构建基因组索引可以作为研究精准高效的基因组序列映射算法的基础,同时为实现多基因组上的序列比对提供研究思路。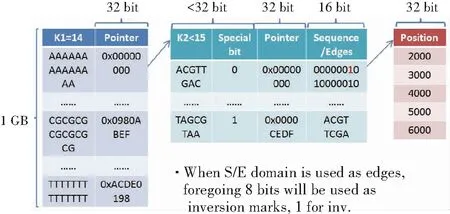
2 索引结构构建方法
2.1 索引的基本组织结构分析
2.2 相似种子合并操作方法
3 结果分析



4 结束语
