微分电化学质谱:研究锂离子电池的一项关键技术
2019-01-04赵志伟彭章泉
赵志伟,彭章泉
微分电化学质谱:研究锂离子电池的一项关键技术
赵志伟1,2,彭章泉1,2
(1中国科学院长春应用化学研究所电分析化学国家重点实验室,吉林 长春 130022;2中国科学技术大学,安徽 合肥 230026)
锂离子电池的安全性问题在很大程度上限制了其在纯电动汽车、规模储能等领域的广泛应用。电池材料|电解质界面副反应所产生的可燃性气体是锂离子电池安全隐患的首要原因。微分电化学质谱是解析锂离子电池产气副反应机制的强有力研究技术。本文综述了微分电化学质谱的发展历程、工作原理、技术要点及其在锂离子电池安全性研究中的应用,并展望了微分电化学质谱在储能领域的机遇、挑战和策略。
微分电化学质谱;锂离子电池;安全
能源存储和转换技术是当今社会实现可持续发展的重要一环。其中,电化学储能系统具有污染少、循环效率高、寿命长及维护成本低等优势而备受关注[1]。研发高比能、高安全性的电化学储能系统是电化学家一直以来追寻的梦想。锂离子电池储能技术以其能量密度高、循环寿命长等优点,具有广阔的应用前景——从便携电子设备到电动车,直至大规模电网储能系统[2]。锂离子电池主要由正极、负极、有机电解液和隔膜组成,通过Li+在正、负极间的可逆嵌入/脱嵌反应实现化学能和电能的相互转换[3],如图1所示。
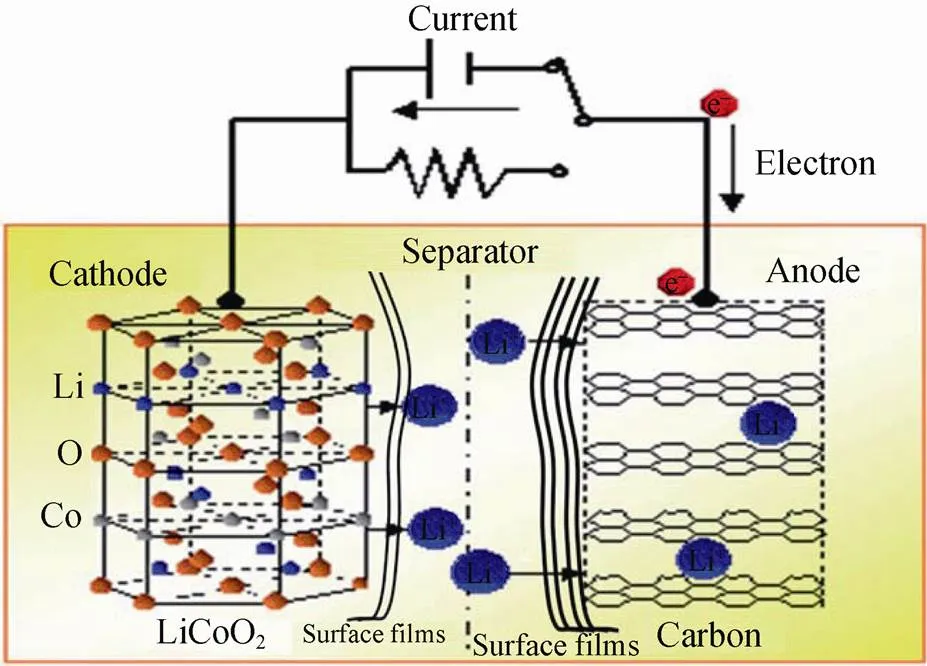
图1 锂离子电池工作原理示意图[3]
在锂离子电池实际工作中,伴随上述可逆主反应,电解液与电极材料亦会发生诸多不可逆的副反应,其中最主要的是固态电解质界面膜(注意它包含正极和负极固态电解质界面膜,这里我们沿用PELED[4]的说法,不做详细的区分,将其统称为SEI膜)的生成[5]。稳定的SEI可以避免电解质和电极材料之间的直接接触,阻止电解质的分解和电极的腐蚀。反之,不稳定的SEI通常反复破裂与重复生长,使得电解质和电极材料持续发生不可逆反应,产生大量可燃性气体。这种情况会造成电池内部压力增加、电池膨胀、电极活性物质的脱落、失火和爆炸等,致使电池存在极大的安全隐患[6]。
理解SEI的形成机制是构筑高性能锂离子电池的关键步骤。传统的非原位手段无法直接从分子水平上解析SEI的形成机制,并且诸多非原位表征技术多在开放的环境下测试,会诱导SEI膜发生物理或化学变化,降低测试准确性。相比于非原位技术手段,原位分析工具可以研究正在进行的物理化学过程、电池不同组件之间的相互作用、从空间和时间上给出SEI膜在形成过程中准确的动态变化信息,大大减小了实验误差[7]。这些原位信息的获取,可以加深我们对锂离子电池SEI膜形成机制的理解,进一步抑制有害过程的发生,对提高锂离子电池储锂性能具有重要的理论指导意义。
先进的原位表征技术,如原位的原子力显微镜(AFM)[8]、X射线衍射(XRD)[9]、扫描电子显微镜(SEM)[10]、红外(FTIR)[11]和拉曼(Raman)[12]光谱等,可以提供关于形貌、结构、成分等有价值的信息,但是它们都不能明晰锂离子电池运行中的产气种类、数量及电极电位之间的关系。这些信息对于理解SEI的形成机制以及提高电池完全性至关重要。伴随着1984年第一台真正意义的微分电化学质谱(differential electrochemical mass spectroscopy,DEMS)[13]问世以及近年来该项技术的发展,为进一步理解锂离子电池的界面问题提供了更多可 能性。
DEMS对于理解锂离子电池SEI形成机制和安全性研究至关重要。因此,本文以DEMS的发展历程、工作原理、技术要点和实验流程为切入点,详细介绍了DEMS技术在锂离子电池中的应用。其次,分析讨论了DEMS和其它互补的原位技术的联用的重要性。最后,对DEMS在储能领域的机遇、挑战和策略进行了展望。
1 发展历程
电化学质谱(electrochemical mass spectroscopy,EMS),是传统电化学与现代质谱分析技术的结合。在名称上,DEMS比EMS多一个D(differential),其有两方面的含义:① DEMS可以测量所产生物种数量的时间导数(derivative),即形成速率;②DEMS采用两级真空泵产生压力差(pressure difference)来输运气体产物。对于DEMS来讲,它最重要部分就是一个“膜”将电池与质谱的真空系统分开,即进样方式。它必须满足两个要求,第一对于挥发物来说是可渗透的,电解液不可渗透。其次,物种从电极界面到质谱检测器的转移时间(响应时间)要足够短。这样,电化学反应的时间或电势和质谱信号才能够完全匹配,实现真正意义的“differential”。因此,本节将以DEMS的进样方式为主线来展现该体系的历史进展,并提出未来发展的方向。
1971年,BRUCKENSTEIN等[14]发明了第一台EMS用于研究亚硝酸根的还原气相产物。他们将电极材料涂覆在Teflon膜上作为多孔电极,电极一侧和电解液接触发生电化学反应,而另一侧通过Tefolon膜连接质谱的真空系统[图2(a)]。这样,电化学反应产生的气相产物通过压力差持续进入一个常规的质谱进行检测,为后来真正的DEMS问世奠定了基础。然而,较厚的电极材料膜大大增加了气相反应产物向质谱扩散的时间,以至于响应时间需要20 s以上。在1984年,WOLTER和HEITBAUM[13]在沿用Teflon膜的基础上,通过改进真空系统,即使用两级真空泵的抽运系统,使得时间常数变得足够短[图2(b),原理参照2.1节],缩短了一半的响应时间。这样,它允许准原位检测电化学反应产物,标志着DEMS的真正问世。
尽管DEMS的Teflon膜进样方式能够避免水分和有机液体等进入质谱,但缓慢的气体转移速度使得DEMS仍然需要10 s以上的响应时间,以及低的气体转移效率(气相反应产物向膜扩散和向电解液扩散的比率)造成了一定的定量误差。随后,为了增加气体转移效率,旋转圆盘电极(RDE)作为DEMS的入口系统[15]被开发[图2(c)]。RDE可以调整转速来控制扩散层厚度,进而达到较高的气体产物转移效率,实现准确定量。1998年,为了进一步提高气体转移效率,开发了薄层电池的进样方式[16][图2(d)]。后来,根据实验目的,基于Teflon膜的不同进样方式也被发展[17]。然而,Teflon膜进样方式本质上决定了部分电化学/化学反应产物没有机会到达质谱(低的气体传递效率)和缓慢的气体转移速度,使得响应时间和定量结果始终不能令人满意,限制了DEMS技术的发展。

图2 电化学质谱进样方式的发展历程
最近10多年,由于储能体系的发展处在一个瓶颈期,特别是锂离子电池,迫切需要DEMS这项技术的突破来理解锂离子电池界面现象,进而推动其发展。通常,电解质在石墨负极上还原分解形成SEI膜,产生大量的气体,使用先前的DEMS系统检测并不困难。然而,与负极和电解液之间的反应相比,正极材料与电解液反应产气量小,之前的DEMS系统已经不再适用。因此,在2007年,NOVAK课题组[18]设计了一个基于“顶空分析”技术和“载气吹扫”进样方式的电化学池[图2(e)]。气态反应产物可连续从电化学电池顶部抽出,通过毛细管进入四极杆质谱仪进行分析。此外,更少电解液用量以及更多电极活性物质的设计,使得检测信号相比于传统体系提高了100倍,可以实现痕量气体的检测。同时,载气吹送进样方式极大地提高了气体的转移速度,使得响应时间进一步缩短。在此基础上,MCCLOSKEY等[19]的Swagelok电化学质谱池设计[图2(f)]进一步减少电解液的用量到40~60mL和125mL的顶部空间,进一步提高检测限到小于1 ppm(1 ppm=10-10)水平。然而,这两种“顶空分析”技术的电化学池设计,并不能完全保证电化学或化学反应所产生的气体100%到达质谱,仍会有少部分气相产物溶解在电解液中,对于定量研究存在一定的误差。此后,为了保证气体传输效率达到或非常接近100%,PENG等[20]的质谱电化学池设计允许螺旋式气体吹扫在电池内部产生压力梯度[图2(g)红线所示路径],以及BREKES等[21]的质谱电化学池设计使得载气流经电解液[图2(h)],这两种“动态”顶空进样方式使得电解液溶解的气体几乎全部进入质谱检测器,实现对反应产生气体的准确定量。
目前来看,载气吹扫进样方式使得气体的转移效率接近100%,定量结果较为准确。然而,DEMS的响应时间(一般在1~5 s)远远不能达到单电子反应水平,比如飞秒级别,这样使得DEMS研究多电子耦合的电化学过程变得尤为困难。此外,用DEMS直接研究实际锂离子电池体系还未见报道,比如圆柱、软包、铝壳电芯等。其中,几个技术难点需要克服:①实际的锂离子体系,如软包电池,化学/电化学产气量比质谱模型电池大很多,这也对DEMS的载气系统势提出了更高要求;②如何将实际的锂离子电池接入质谱的载气系统;③气体定量准确性如何提高。未来的研究方向将更多集中在提高DEMS的时间分辨率,尽量缩短DEMS气路长度和毛细管管径以及尽可能增加载气流速或设计新型的进样方式。另外,设计新的接近实际电池体系的质谱电化学池和载气系统也至关重要,这对实际工业生产具有重要意义。
2 DEMS体系
图3展示了本文作者课题组DEMS系统的组成。主要部件是气体净化装置、流量控制器、电池单元、三通阀、可控温冷阱、电解液过滤膜和PEEK管线。本节将着重介绍DEMS系统的组成、原理、实验流程、实验注意事项以及可能的误差等。
2.1 系统构成
2.1.1 载气系统
如图3(a)和3(c)所示,载气首先由气体钢瓶进入气体净化装置,排除杂质气体(主要是H2O和CO2等)对实验的干扰,然后进入流量控制器。根据实验体系的不同,质量流量控制器的流量设定通常为0.1~2 mL /min(注:流速太慢容易降低气相产物的转移效率,流速太快使得电解液挥发加剧)。之后,载气流经电池单元,若将阀门a和b通过线路c连通,则可清洗整个系统;如果阀门a和b通过线路d和e连通,将电化学/化学反应产生的气体几乎全部强制“带入”载气中,除了这些,一些易挥发的有机溶液也容易进入载气中。流经电池单元的载气依次通过冷阱和Teflon膜过滤单元后,载气中的有机电解液被过滤,电化学/化学反应产生的气体进入质谱进行分析检测。
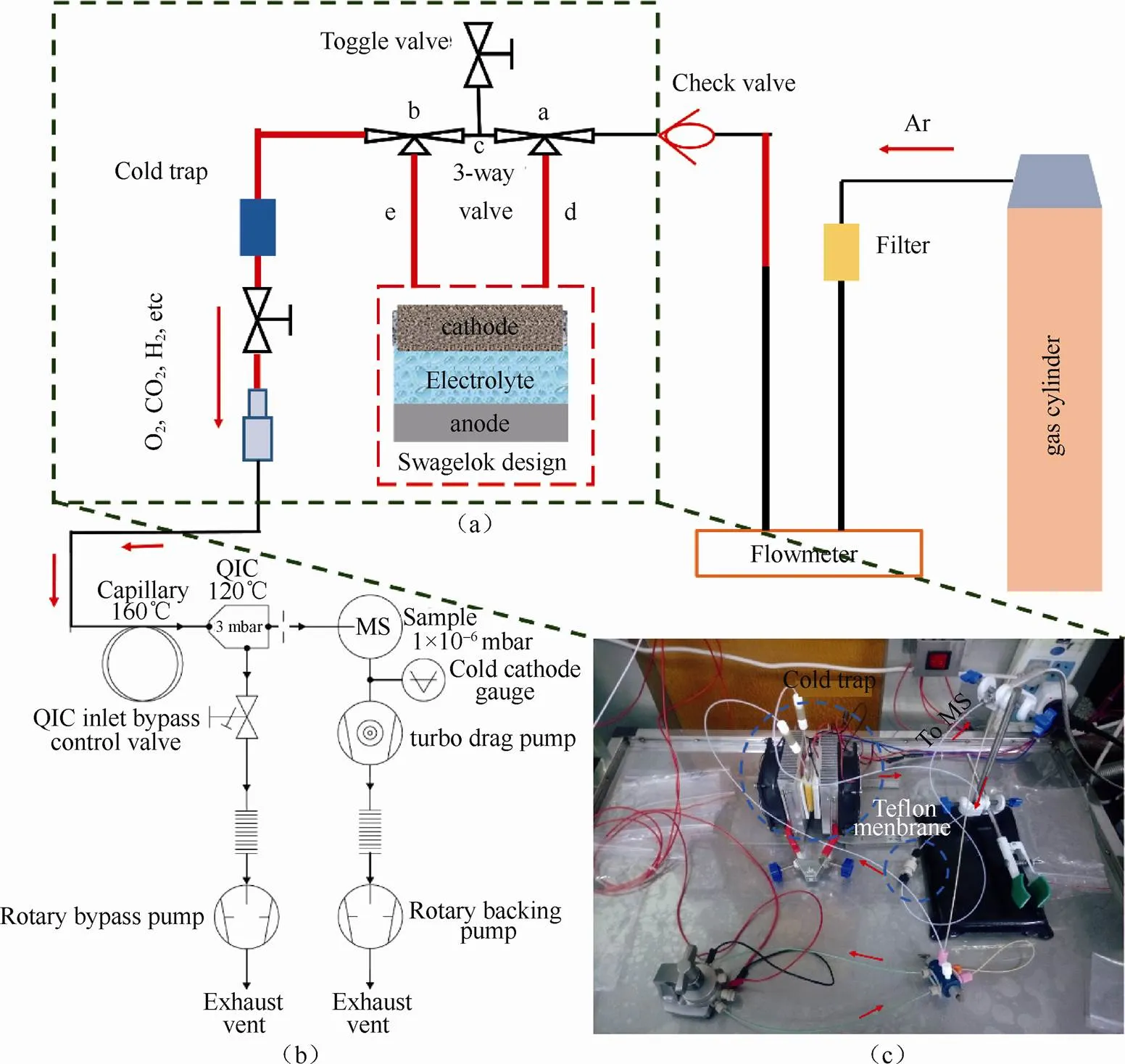
图3 (a)DEMS示意图;(b)质谱真空系统示意图;(c)载气系统和电化学反应装置数码照片
对于DEMS的吹扫进样方式,高纯度的惰性载气是必需的。其次,载气不能和电池组件发生反应,比如N2与电池中的金属锂发生反应。尽管Ar经常作为载气,但它其实并不是一个好的选择。Ar会缩短质谱仪检测器的使用寿命,而且它会电离出18、20、36以及40的质荷比碎片,可能会与分析的气体重叠。综合考虑,高纯度氦气作为载气是一个不错的选择[21]。
2.1.2 真空系统
不管早期的Teflon膜进样或现代载气吹扫进样方式,其基本原理一致[22]。载气携带的电化学/化学反应气相产物以(单位mbar·L/s, 1 bar=105Pa)的流量进入质谱,静态条件下,通过真空泵将其输运并满足如式(1)
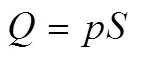
式中,是质谱离子室的压力,一般小于10-3mbar;是泵速,L/s。一个物种的检测时间常数由离子室体积0和泵速决定,满足式(2)

在非静态条件下,突然的流量变化,会使得压力改变,即
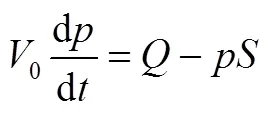
求解后,得到式(4)
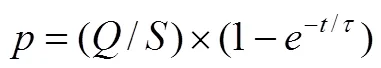
根据式(1),离子室的压力一定,当流量比较大时,需要较大的泵速。若0=1 L和= 200 L/s时,可达到一个较短的时间常数(5 ms)。而对于质谱分析室而言,必须用单独的涡轮分子泵,以使压力低至10-5mbar,一个50 L/s左右的涡轮分子泵可以满足要求。这时,离子室和分析室之间存在一个压力差,并且使用了两个涡轮分子泵使得复杂性增加。为了减小其影响,可以在分析物进入质谱的真空系统之前连接一个转速较低的旋转泵来泵送,大大减小了和值。在这种情况下,离子室放置在一个独立的空间内,并且与质谱的分析室一起被一个50 L/s的简单涡轮分子泵泵送[图3(b)]。这样,不仅简化了质谱的真空系统,而且方便连接商用的进样系统。然而,一些物种可能在该压力下发生 吸附或冷凝,因此需要进行加热毛细管进样,比如160 ℃。
2.1.3 电池单元
相对于常规的锂离子测试电池,电化学质谱池的设计需要额外的进出气口,并且气密性需要严格保证。它的核心主要是气路设计,因为这决定了气体传输效率和质谱定量的准确性。对于图4(a)中的气路设计,电化学/化学反应产生的气体依靠压力差(0-1)扩散到载气中。这样的设计使得少部分气体不能随载气到质谱中,而且同时形成一定的涡流,降低气体传输效率。此外,气体进入载气中使得载气压力突然(2>1)增加,这在一定程度上也会影响测试结果的准确性。在图4(b)中,载气流进电解液组分,将电化学/化学反应产生的气体从电解液带入载气中。尽管这样的设计提高了气体的传输效率,但是也使得挥发性电解液大量进入载气中。如果测试时间过长,电解液会被耗干而迫使测试终止。另外,载气进入电解液会形成大量的涡流,可能使得电池系统不能趋于稳定状态。针对以上2种电池设计的弊端,图4(c)的电池气路设计,在电池上方形成螺旋梯度气体流场[见图2(g)三维示意图],使得气流更加稳定,大大减小了涡流情况的发生概率。此外,螺旋梯度流场也能增加气路长度,缓建载气压力的骤然变化而且提高了气体传输效率。
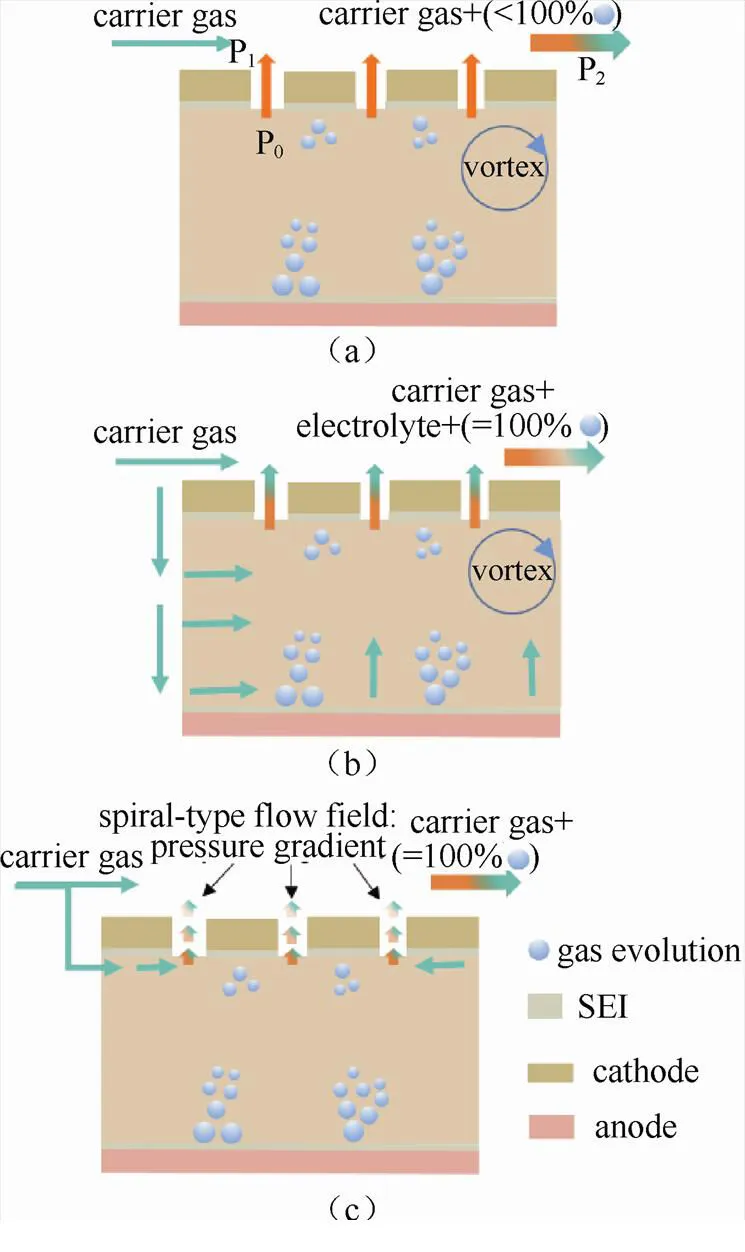
图4 不同的电化学质谱池气路示意图
2.1.4 其它组件
DEMS对痕量的污染物非常敏感,各组分的清洁度对于研究结果的准确性有着重要影响。
第一,选择合适的连接管和接头。若其中含有聚四氟乙烯(PTFE)部件,它们的使用在高纯度气体管线系统中不是优选的,因为电解质/蒸汽可能会溶解PTFE(时间长的情况下)。因此,通常选用PEEK材质的连接管和接头等。
第二,为了保护质谱仪免受溶剂组分的影响并延长其使用寿命,电池单元和质谱间接入自制的PID温度调节的冷阱,作为进样口的第一重保护。温度调节方面,需要根据电解质溶液的冰点进行调节,对于常规的锂离子的碳酸酯类电解液,冷阱温度通常设定为-25 ℃。该温度不仅保护质谱仪,而且提高了质谱仪的检测准确度。一般来说,挥发性电解质组分容易被电离,导致目标检测物质的电离概率减少。此外,进入到质谱的电解质也会产生碎片,与可能的反应产物重叠(如CO、CO2、C2H4等),干扰测试信息。为此,再加入Teflon膜过滤装置,过滤可能没有冷却的电解液蒸汽,成为进样口第二重必要保护。
2.2 实验流程
首先,在DEMS研究锂离子电池副反应过程中,任何痕量杂质(如水、HF及少量空气等)都可能参与化学或电化学反应,影响测试结果的准确性。因此,电解质盐和溶剂必须在使用前进行纯化和干燥(除非这些试剂的纯度是电池级),以保证污染程度最小化。
电解质锂盐应具有高纯度,一般需要达到99.99%以上。对于纯度较低的电解质盐,重结晶过程是必不可少的一个步骤。对于达到电池级的锂盐,比如LiClO4和LiPF6不需要重结晶,但也需要在 120 ℃真空干燥24 h才可以使用。
溶剂的种类和纯度与锂离子电池SEI膜的形成、锂离子的电导率、电池的安全性等有直接的关系。有机溶剂需要在配制电解液前进一步纯化和除水。对于高沸点溶剂,溶剂的纯化分为三个步骤。首先,将5 g氢化钙(CaH2)加入到含有700 mL溶剂的圆底烧瓶中,并将溶剂在室温下搅拌至少 24 h以预先脱水。预脱水后,溶剂在真空密闭的条件下进行减压蒸馏。然后,将蒸馏溶剂直接从蒸馏装置中取出(不能暴露于空气),并转移到氩充满的手套箱中。最后,用新鲜活化的3 Å(1 Å=10-10m)分子筛进一步干燥蒸馏过的溶剂,这个过程至少需要3天以上。对于低沸点溶剂,纯化过程和高沸点溶剂相似,不同的是常压蒸馏即可。需要注意的是整个过程也应该在氩气氛下以避免空气污染。最终,纯化后的溶剂的水含量需要通过Mettler-Toledo Karl Fischer滴定仪测定,其最终含水量应小于10 ppm。
此外,购买的分子筛在使用前也需要干燥或活化。首先用乙醇洗涤分子筛,在150 ℃烘箱中干燥12 h,然后置于干燥管中,并在300 ℃下用Büchi在真空下进一步干燥24 h。干燥完毕后,在不暴 露于空气的情况下将分子筛转移到Ar充满的手套箱中。
第二,正负极的极片制作可参照文献[23]。需要注意的是,在小电流充放电或电极活性物质负载量较少的情况下,气相产物相对较少,使得质谱的信号可能不明显。经验来说,Swagelok电化学质谱池所需极片直径为24 mm,活性物质负载量一般在5 mg以上,需要50 mA/g以上的充放电电流。
第三,Swagelok电化学质谱电池的组装和正常电池一致,组装后接入DEMS系统[图3(c)]。首先将电池短接,载气排干净DEMS系统中的空气后(大约1 h),再将电池接入载气系统。待基线平稳后(大约1 h),便可以对电池充电(或放电,取决于你的研究问题,此处指正常的锂离子电池)。充电完成后,需要基线再次平稳后,才可对电池继续放电。
2.3 电化学方法选择
在锂离子电池工作过程中,恒定电压和恒定电流是两种常用的充放电方法。对于恒定电压模式,在充电初期充电电流会比较大,有可能造成电池温度上升过快,对电池体系产生不利影响;对于恒定电流模式,在后期可能出现充电电流超过可接受电流而导致电池电解液发生副反应(如析气反应)。若恒定电流太小,延长充电时间,降低充电效率,而且产气量太小可能会低于DEMS的检测限,因此,在DEMS实验中,需要选择合适的电化学方法以及参数(电流、电压)才能得到理想的实验结果。
电化学反应速率取决于电极电位,循环伏安法(CV)已经被广泛应用于快速、定性和定量地给出电化学反应电位之间的关系。通常,循环伏安技术(经验地扫速0.1~1.0 mV/s), 可以确定电流密度,电位与消耗或释放气体速率之间的粗略关系。根据这个关系,较容易获得较为合适的电流密度或电位。这样,就可以在较为理想的条件下(减少了不必要的副反应)研究锂离子电池系统。当然,这里不包括极端条件(高过电势,高电流密度等)下的DEMS研究。但是,由于实验条件、设备、电池系统等的不同,参数的选择往往只能依靠经验,我们也只能提供粗略的经验方法来确定实验参数。另外,还可以利用其它方法(变流和变压充放电等),这取决于研究问题。
2.4 数据处理
首先是流量计的校准。通常,流量计是基于某种气体校准,比如N2。当DEMS载气为非N2时需要校准载气流量。以载气Ar作为例子,流量计显示流量为m,质谱输出信号为signal(一般为体积比)时,实际的Ar流量为
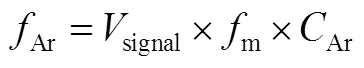
式中,Ar为Ar校正因子。当需要研究一些气体对锂离子电池的影响时,即载气为混合气时也需要对载气流量校准。我们这里以Ar和O2的体积 比为8∶2为例,首先,校正因子mix被计算根据如式(6)

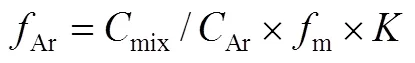
式中,m是流量计的流量,是Ar在混合气中的比例(这里,=0.8)。

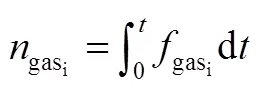


2.5 误差分析
我们首先介绍DEMS的基本定量原理[22],便于更好的理解整个数据分析过程中存在的误差。通常,质谱通过测定相应物种的离子电流来记录物种的形成速率。质谱测定物种i的离子电流i和进入质谱物种i的流量i(mol/s)成正比,满足式(10)
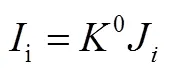
式中,0包含质谱仪的所有设置和物种i的电离概率。当电化学反应产生物种i时,i可根据如下公式计算
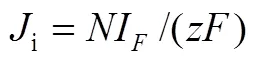
式中,是电化学反应的电子转移数,是法拉第常数,F是法拉第电流,是转移效率,即电化学反应的气相产物到质谱检测器的比率(通常<1,部分气相产物会扩散到电解液中)。将式(10)和(11)整合,可得
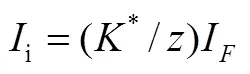

使用已知的电化学反应(比如析氢反应)校准时,*以及0很容易被获得。这样,我们很容易得到电化学反应产生物种i的形成速率。
对于载气吹扫方式进样的DEMS,校正过程变得相对简单,也更准确。根据式(10),只需要知道载气的流量和相应的质谱电流信号,便可得出0系数,校正过程更简洁。准确性主要体现在:①电化学反应产生的气相物种几乎可以全部到达质谱检测器,即≈1;②不必考虑校正过程双电层电容对法拉第电流的影响。存在的误差可能有:①载气和需要检测的气体的电离概率不同;②当载气中混入电化学反应产生的气体时,载气流量与实际存在偏差。由于电化学/化学反应过程中产生的气体量很小,这些误差几乎可以忽略。
其次数据处理过程可能存在误差。由于较大信噪比的存在,数据处理时会采用不同的基线扣除和数据平滑方式。这里以图5作为一个例子,当数据不采用任何手段处理时,计算所得的CO2释放量为0.286 nmol,若采用Savitzky-Golay方法做平滑处理,CO2释放量则发生了一些变化。若DEMS的信号波动(峰)更多或信号相对于基线不明显时,平滑处理会引入更多的误差。因此,在数据处理时尽量遵循原始数据,减少人为干扰,这样方便不同的已发表文章的数据对比。
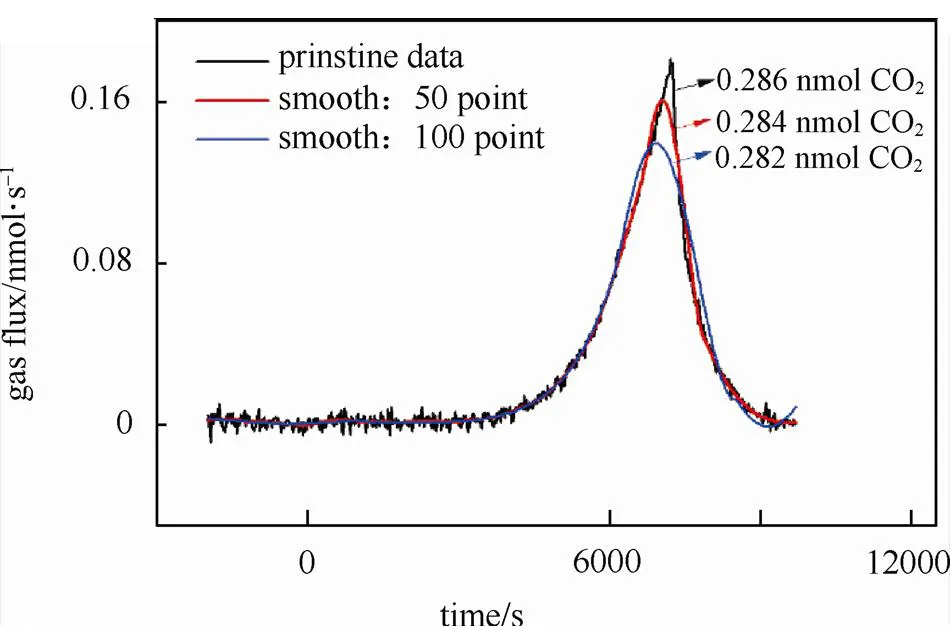
图5 DEMS分析CO2释放在1 mol/L LiTFSI-TEGDME电解液体系
3 DEMS在锂离子电池中的应用
锂电池的组件,包括正极、负极、电解液和隔膜等对电池的性能影响至关重要。尽管目前已经有大量先进的表征技术对锂电池进行了系统研究,提供了丰富的界面和物质结构变化等信息,但是它们都不可能原位检测并定量锂电池随电势变化的气体产物,以及电池的动力学信息。DEMS作为锂离子电池产气分析和安全性问题研究的强有力技术,它的研究进展已经在许多化学类顶级刊物上被报 道[7,24-26]。因此,本节重点强调DEMS如何用来研究锂电池的各个组件,如何根据DEMS的结果推断反应机制,为初学者提供研究思路。
3.1 电解液
丙烯碳酸酯(propylene carbonate,PC)作为一种锂离子电池广泛使用的电解液,几种可能的分解机制被提出。第一种可能分解机制是PC氧化开环形成丙酮或丙醛,伴随着CO2释放[27]。另外一种分解机制认为PC分解产生CO2,丙醛以及顺式和反式-2-乙基-4-甲基-1,3-二氧戊环[28]。接下来,以文献[29]为例子,我们将解释如何用DEMS来验证PC可能的反应机制。
在DEMS测量中,根据可能的反应机制,首先要选择可能的电解液分解产物的质量数,通过扫描全谱(/= 0~300)得到。在所得质谱信号中寻找与离子电流或电位相关的质量数,独立的信号,即与离子电流和电位无关,便可以从反应机制中被排除。正如图6(a)所展示,质量数58是与电位相关的信号。因此,它可以排除乙烷、乙烯、丙烷和丙烯的存在。这也就从实验角度验证了PC分解产生丙酮的分解机制[29]。
图6 (a)离子电流随电位的变化曲线[29];(b)LNMO/石墨电池和LNMO/LixFe1-xPO4电池(c)充放电曲线以及相应的DEMS信号[30]
此外,DEMS在鉴别电解液的“好坏”上也具有一定的优势。这里,一个经典例子[31]来介绍同位素标记与DEMS技术结合在锂离子电池产气来源及比率分析的应用。
锂离子电池在储存过程中不可避免地会产生一些气体,CO2和CO等最为普遍。为了评估电池各组件对产生气体的贡献,ONUKI等[31]使用13C标记的EC或DEC作为电解液。实验发现,13C-EC /13C-DEC作为电解液时,产生的13CO2比例为63%。当使用13C-EC/12C-DEC电解液时,产生的13CO2为52%。因此,他们推断EC、DEC和非电解液部分贡献的CO2分别为52%、11%和37%。进一步,他们使用单正极或负极放于电解液中来评估其产气比例,表明正极是12CO2的主要贡献者,负极只会产生少量的12CO2。因此,降低电解液中EC的含量和正极材料中碳酸盐杂质可减少锂离子电池产气量,提高安全性能。不受限于这个例子,DEMS-同位素标记技术还可以通过标记锂离子电池组件正极的氧元素以及溶剂和电解质的碳元素等,研究正极材料的稳定性以及电解质和溶剂的分解机制,相关的工作亟需开展。
3.2 正极/负极-电解液界面
目前,锂离子电池中最具前景的正极材料是LiNi0.5Mn1.5O4(LNMO),最常用的负极材料是石墨。然而,它们的应用仍然存在一些限制,其中之一是容量随循环而衰减,主要是由于过渡金属溶解和正极或负极侧的电解质分解。理解LNMO/石墨电池的反应机制,将会支持锂离子电池系统的进一步发展。本节以DEMS作为原位分析工具,分析讨论 LNMO/石墨电池的反应机制。

为了验证以上推断,并将正极上的气体释放反应与负极侧发生的气体释放反应分离开来,需要组装LNMO/LiFe1-PO4或LiFe1-PO4/石墨半电池。与LNMO/石墨系统相比,LNMO/LiFe1-PO4电池总的压力增加小得多。此外,CO2是主要的气体产物,C2H4和H2都没有被检测到,因此,C2H4和H2实际上主要是在石墨负极一侧产生的。此外,在LiFe1-PO4/石墨电池上的DEMS表明,在首圈充电过程没有二氧化碳的释放,而是生成了C2H4和H2。综上,CO2主要是在正极由于氧化电解液分解产生的,而C2H4和H2主要与石墨负极SEI膜形成相关。
3.3 添加剂
除了正负极和电解液之外,电解液添加剂对于改善锂离子电池寿命至关重要。在首次充电过程中,添加剂可优先于电解质分解,形成稳定的SEI层,极大较少了电解质的消耗以及可燃性气体的释放。本节将以DEMS为例,来说明它在甄别添加剂方面的应用。
碳酸亚乙烯酯(vinylene carbonate, VC)由于较好的热稳定性,是目前研究最广泛的锂离子电解质添加剂之一[32]。首先,组装石墨/LiFe1-PO4电池(若研究Li负极,则组装Li/LiFe1-PO4电池)。然后,执行空白试验,当没有添加剂时,检测前几圈(SEI形成通常在前几圈)释放气体种类(如C2H4、CO2、H2等)并定量,计算气体释放速率。最后,电解液加入不同含量VC(一般要低于5%)时,观察气体释放含量是否减少,如使用EC电解液时,通常观察C2H4是否减少。当气体释放总量以及释放速率最小时,得出最佳的添加剂使用比例。
总之,由锂离子电池产气而导致的安全问题始终限制着它的广泛利用。减缓或避免锂离子电池产气具有重要的实际意义。DEMS在鉴别以及理解气体产生机制具有不可替代作用:①电解液溶剂或盐的分解机制;②负极和电解液之间的反应原理,即形成SEI膜的过程;③正极材料稳定性。最终,我们才可以“对症下药”,进一步实现安全和高比能的锂离子电池的目标。
4 DEMS与其它先进技术的联用
电解质和电极之间的界面(主要是SEI),保持健康性对电池性能和安全性方面起着极其重要的作用。原位分析能够判断电池的健康状况以及诊断电池问题的根源。如前文所述,DEMS可以监测和识别和计算由于界面反应而产生的气态物质。然而,DEMS单一分析技术不能提供一次理解界面现象的所有所需信息,如界面的组成、形貌、力学和电学性质。如果用不止一种原位技术分析电池体系,即分批处理,可能会导致某些人为处理错误,并使处理时间耗费。因此,将各种原位工具集成在一起有助于对锂电池界面进行全面有效分析(图7)。不幸的是,由于目前的技术限制,多种原位分析方法集于一体的电化学池设计鲜有报道。下文将介绍几种与DEMS具有良好互补效应原位技术。

图7 电极-电解质界面集成分析工具
SEI的组成决定了界面层的机械稳定性和离子迁移的电化学性质。原位XRD已被用于识别存在于界面层中的无机物质。然而,迄今为止,只有LiF、LiOH等能被X射线源检测到,而其它SEI组分由于其非常小的微晶尺寸而很难被探测到。纳米级厚度的SEI和小尺寸的无机成分限制了XRD的应用。与XRD相比,基于X射线吸收的光谱(XAS[33]),如XANES[34]和EXAFS[35]技术,被认为更适合研究界面组成。它对极低浓度的元素异常敏感。XAS不需要长程有序,可用于非晶态分析以及小纳米粒子。此外,X射线反射技术(XRR)[36]也可用于确定SEI密度、厚度和粗糙度,但是它需要相当平滑的电极表面进行分析,而在使用复合结构的实际电池电极中情况并非如此。X射线光电子能谱分析(XPS)[37]具有精确分析SEI化学成分的能力,然而高真空的操作条件,使得XPS原位分析仍然是一个挑战。
FTIR[38]和Raman[39]被用于互补性鉴定SEI的有机和无机成分,并且这两种技术已被证明是电池界面和电解质在分子水平上分析的快速和非破坏性分析工具。然而,这些技术只能识别遵循其特定光谱选择规则,并且在特定波长具有振动模式的物种。NMR[40]等基于磁力的技术也可监测SEI生长过程。然而,NMR信号质量很差,受到限制。随着NMR和MRI[41]成像技术的发展,可用来研究锂离子在电池的空间浓度梯度变化。最近报道的TOF-SIMS[42]技术可以原位监测不同离子的浓度变化,生成各组分分布的3D图。
SEI表面形态变化和物理化学性质反映了SEI的机械稳定性和稳健性。不同类型的电极和电解质形成的SEI千差万别。有几种分析工具用于获得关于SEI形态的信息,如SEM[42]、TEM[43]和AFM[44]等,具有微米级到纳米级的分辨率。基于氮化硅探针的AFM可以用来分析SEI表面,而不用考虑其导电性。AFM对于研究界面生长非常准确,并且适合于随着原位监测SEI生长过程和厚度变化。与基于探针成像类似,SEM和TEM也适用于纳米级的形态分析。然而,由于高真空和电子束的腐蚀性,对于使用SEM和TEM的电池电解质原位电子成像是一项巨大的挑战。此外,AFM技术还可以估计电极界面处产生的应力和杨氏模量,进而确定SEI物理化学性质。
目前为止,仅有少数报道将几种原位分析技术整合到一起。比如,JUSYS等[45]在单个电化学池中对DEMS和电化学石英晶体微天平(EQCM)进行了整合,实现了电极反应气相产物和固体或可溶性产物的同时检测。美国加州理工学院的ANDERSON[46]在传统的AFM尖端涂覆了金纳米粒子,并用它成功地将形貌信息和Raman的化学组合信息整合在一起。集成多种原位技术于一体的工作亟需开展。此外,现有分析工具的空间和时间分辨率不匹配以及高功率或长时间的辐射,对SEI的原位研究也带来了挑战。冷冻电子显微镜[47]的发展可以最小化光束损伤,将可能会是研究SEI的一种有力工具。
5 结 语
在锂离子电池领域,电解液/电极界面的稳定性对Li+传输,电池动力学以及安全性至关重要。大量的工作集中在开发正负极材料,电解液和添加剂等方面,企图寻求稳定的SEI层,进而改善电池性能。然而,这些盲目的SEI层设计费时费力。因此,构建令人信服的SEI演化机制(电极和电解液如何相互作用)是设计稳健的电池储能体系的必要步骤。界面的研究不仅有助于改进现有的锂离子电池,而且通过构建稳固的界面还可实现高比能的锂空气和锂硫电池的商业化。
为了理解SEI演化机制,原位分析技术至关重要。我们不仅强调DEMS原位监测、识别以及定量界面反应而产生的气态物质,而且强调使用与DEMS互补的技术来共同理解SEI的动态演化机制(图7)。通常,每种原位技术都有相应的电池设计,其都有一定的优点和局限性。例如,DEMS研究使用的Swagelock电池是不透明的腔体,这限制了DEMS和原位光谱技术的集成。此外,各种原位分析手段都可能或多或少破坏界面的实际化学或物理条件,比如DEMS改变了电池系统的压力,光谱技术的光源损坏界面结构等。最重要的是,仪器检测限和谱图采集的时间跨度对获取合理的数据有很大的影响。因此,未来的研究方向之一需要集中在合理的电池模型设计,可以高度集成各种原位分析手段,同时可提高检测限以及减少对电池体系的影响。
此外,大多数工作集中于模型电极,比如原位Raman研究所使用的Cu、Ag、Au等。这可能与实际电极条件下界面形成不同。而且,SEI的不稳定性质在应用一些破坏性技术(如XPS)时也存在问题。此外,在分子水平上的某些研究,如Li+的嵌入和溶剂化,分子分解形成界面层的热力学等这些问题都未能被原位技术完美解决。因此,未来的SEI界面的研究需要通过各种理论计算来补充验证。
尽管DEMS提供了锂离子电池界面反应气体相关的独特信息,但我们并不否认其它原位工具在研究SEI界面的重要作用。开发DEMS以及与DEMS集成互补的联用技术将有利于深入剖析锂离子电池的界面反应机制和其它电化学器件的微观结构研究,如锂空气电池、锂硫电池、锌空气电池、镁电池和钠电池等。
[1] YANG Z, ZHANG J, KINTNER-MEYER M C W, et al. Electrochemical energy storage for green grid[J]. Chemical Reviews, 2011, 111: 3577-3613.
[2] NITTA N, WU F, LEE J T, et al. Li-ion battery materials: Present and future[J]. Materials Today, 2015, 18: 252-264.
[3] ETACHERI V, MAROM R, RAN E, et al. Challenges in the development of advanced Li-ion batteries: A review[J]. Energy & Environmental Science, 2011, 4: 3243-3262.
[4] PELED E. The electrochemical behavior of alkali and alkaline earth metals in nonaqueous battery systems—The solid electrolyte interphase model[J]. Journal of the Electrochemical Society, 1979, 126: 2047-2051.
[5] VERMA P, MAIRE P, NOVÁK P. A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries[J]. Electrochimica Acta, 2010, 55: 6332-6341.
[6] AURBACH D, MARKOVSKY B, SALITRA G, et al. Review on electrode-electrolyte solution interactions, related to cathode materials for Li-ion batteries[J]. Journal of Power Sources, 2007, 165: 491-499.
[7] TRIPATHI A M, SU W N, HWANG B J. In situ analytical techniques for battery interface analysis[J]. Chemical Society Reviews, 2018, 47: 736-851.
[8] TOKRANOV A, SHELDON B W, LI C, et al. In situ atomic force microscopy study of initial solid electrolyte interphase formation on silicon electrodes for Li-ion batteries[J]. ACS Applied Materials & Interfaces, 2014, 6: 6672-6686.
[9] HATCHARD T D, DAHN J R. In situ XRD and electrochemical study of the reaction of lithium with amorphous silicon[J]. Journal of the Electrochemical Society, 2004, 151: A838-A842.
[10] GUERFI A, DONTIGNY M, CHAREST P, et al. Improved electrolytes for Li-ion batteries: Mixtures of ionic liquid and organic electrolyte with enhanced safety and electrochemical performance[J]. Journal of Power Sources, 2010, 195: 845-852.
[11] MOSHKOVICH M, COJOCARU M, GOTTLIEB H E, et al. The study of the anodic stability of alkyl carbonate solutions by in situ FTIR spectroscopy, EQCM, NMR and MS[J]. Journal of Electroanalytical Chemistry, 2001, 497: 84-96.
[12] HY S, FELIX F, RICK J, et al. Direct in situ observation of Li2O evolution on Li-rich high-capacity cathode material, Li[NiLi(1-2x)/3Mn(2-)/3]O2(0≤≤0.5)[J]. Journal of the American Chemical Society, 2014, 136: 999-1007.
[13] WOLTER O, HEITBAUM J. Differential electrochemical mass spectroscopy (DEMS)—A new method for the study of electrode processes[J]. Berichte der Bunsengesellschaft für Physikalische Chemie, 1984, 88: 2-6.
[14] BRUCKENSTEIN S, GADDE R R. Use of a porous electrode for in situ mass spectrometric determination of volatile electrode reaction products[J]. Journal of Materials Chemistry, 1971, 18: 5638-5646.
[15] TEGTMEYER D, HEITBAUM J, HEINDRICHS A. Electrochemical on line mass spectrometry on a rotating electrode inlet system[J]. Berichte der Bunsengesellschaft für Physikalische Chemie, 1989, 93: 201-206.
[16] BOGDANOFF P, FRIEBE P, ALONSO-VANTE N. A new inlet system for differential electrochemical mass spectroscopy applied to the photocorrosion of p-InP(111) single crystals[J]. Journal of the Electrochemical Society, 1998, 145: 576-582.
[17] GAO Y, TSUJI H, HATTORI H, et al. New on-line mass spectrometer system designed for platinum-single crystal electrode and electroreduction of acetylene[J]. Journal of Electroanalytical Chemistry, 1994, 372: 195-200.
[18] HOLZAPFEL M, WÜRSIG A, SCHEIFELE W, et al. Oxygen, hydrogen, ethylene and CO2development in lithium-ion batteries[J]. Journal of Power Sources, 2007, 174: 1156-1160.
[19] MCCLOSKEY B D, BETHUNE D S, SHELBY R M, et al. Solvents' critical role in nonaqueous lithium-oxygen battery electrochemistry[J]. The Journal of Physical Chemistry Letters, 2011, 2: 1161-1166.
[20] PENG Z, FREUNBERGER S A, CHEN Y, et al. A reversible and higher-rate Li-O2battery[J]. Science, 2012, 337: 563-566.
[21] BERKES B B, JOZWIUK A, VRAČAR M, et al. Online continuous flow differential electrochemical mass spectrometry with a realistic battery setup for high-precision, long-term cycling tests[J]. Analytical Chemistry, 2015, 87: 5878-83.
[22] BALTRUSCHAT H. Differential electrochemical mass spectrometry[J]. Journal of the American Society for Mass Spectrometry, 2004, 15: 1693-1706.
[23] 王其钰, 褚赓, 张杰男, 等. 锂离子扣式电池的组装,充放电测量和数据分析[J]. 储能科学与技术, 2018, 7(2): 324-344.
WANG Qiyu, CHU Geng, ZHANG Jienan, et al. The assembly, charge-discharge performance measurement and data analysis of lithium-ion button cell[J]. Energy Storage Science and Thechnology, 2018, 7(2): 324-344.
[24] XU K. Electrolytes and interphases in Li-ion batteries and beyond[J]. Chemical Reviews, 2014, 114: 11503-11618.
[25] GAUTHIER M, CARNEY T J, GRIMAUD A, et al. Electrode-electrolyte interface in Li-ion batteries: Current understanding and new insights[J]. The Journal of Physical Chemistry Letters, 2015, 6: 4653-4672.
[26] NOVÁK P, GOERS D, HARDWICK L, et al. Advanced in situ characterization methods applied to carbonaceous materials[J]. Journal of Power Sources, 2005, 146: 15-20.
[27] JOHO F, NOVÁK P. SNIFTIRS investigation of the oxidative decomposition of organic-carbonate-based electrolytes for lithium-ion cells[J]. Electrochimica Acta, 2000, 45: 3589-3599.
[28] ARAKAWA M, YAMAKI J I. Anodic oxidation of propylene carbonate and ethylene carbonate on graphite electrodes[J]. Journal of Power Sources, 1995, 54: 250-254.
[29] UFHEIL J, WÜRSIG A, SCHNEIDER O D, et al. Acetone as oxidative decomposition product in propylene carbonate containing battery electrolyte[J]. Electrochemistry Communications, 2005, 7: 1380-1384.
[30] MICHALAK B, BERKES B B, SOMMER H, et al. Gas evolution in LiNi0.5Mn1.5O4/graphite cells studied in operando by a combination of differential electrochemical mass spectrometry, neutron imaging, and pressure measurements[J]. Analytical Chemistry, 2016, 88: 2877-2883.
[31] ONUKI M, KINOSHITA S, SAKATA Y, et al. Identification of the source of evolved gas in Li-ion batteries using C-labeled solvents[J]. Journal of the Electrochemical Society, 2008, 155: A794-A797.
[32] ZHANG B, METZGER M, SOLCHENBACH S, et al. Role of 1,3-propane sultone and vinylene carbonate in solid electrolyte interface formation and gas generation[J]. The Journal of Physical Chemistry C, 2015, 119: 11337-11348.
[33] SHEARING P, WU Y, HARRIS S J, et al. In situ X-Ray spectroscopy and imaging of battery materials[J]. Electrochemical Society Interface, 2011, 20: 43-47.
[34] HU E, YU X, LIN R, et al. Evolution of redox couples in Li- and Mn-rich cathode materials and mitigation of voltage fade by reducing oxygen release[J]. Nature Energy, 2018, 3: 690-698.
[35] DING Y, LI Z F, TIMOFEEVA E V, et al. In situ EXAFS-derived mechanism of highly reversible tin phosphide/graphite composite anode for Li-ion batteries[J]. Advanced Energy Materials, 2018, 8: doi: 10.1002/aenm.201702134.
[36] YOU H, PIERCE M, KOMANICKY V, et al. Study of electrode surface dynamics using coherent surface X-ray scattering[J]. Electrochimica Acta, 2012, 82: 570-575.
[37] DEDRYVÈRE R, LARUELLE S, GRUGEON S, et al. XPS identification of the organic and inorganic components of the electrode/electrolyte interface formed on a metallic cathode[J]. Journal of the Electrochemical Society, 2005, 152: A689-A696.
[38] JUNFENG Y, NICKOLAY S, ALEXANDER K, et al. In-situ spectro-electrochemical insight revealing distinctive silicon anode solid electrolyte interphase formation in a lithium-ion battery[J]. Chemistry Select, 2016, 1: 572-576.
[39] LI G, LI H, MO Y, et al. Further identification to the SEI film on Ag electrode in lithium batteries by surface enhanced Raman scattering (SERS)[J]. Journal of Power Sources, 2002, 104: 190-194.
[40] MEYER B M, LEIFER N, SAKAMOTO S, et al. High field multinuclear NMR investigation of the SEI layer in lithium rechargeable batteries[J]. Electrochemical and Solid-State Letters, 2005, 8: A145-A148.
[41] TANG W, GOH B M, HU M Y, et al. In situ Raman and nuclear magnetic resonance study of trapped lithium in the solid electrolyte interface of reduced graphene oxide[J]. The Journal of Physical Chemistry C, 2016, 120: 2600-2608.
[42] DUPRÉ N, MOREAU P, DE VITO E, et al. Multiprobe study of the solid electrolyte interphase on silicon-based electrodes in full-cell configuration[J]. Chemistry of Materials, 2016, 28: 2557-2572.
[43] KINOSHITA K, BONEVICH J, SONG X, et al. Transmission electron microscopy of carbons for lithium intercalation[J]. Solid State Ionics, 1996, 86-88: 1343-1350.
[44] BECKER C R, PROKES S M, LOVE C T. Enhanced lithiation cycle stability of ALD-coated confined a-Si microstructures determined using in situ AFM[J]. ACS Applied Materials & Interfaces, 2016, 8: 530-537.
[45] JUSYS Z. A new approach for simultaneous DEMS and EQCM: Electrooxidation of adsorbed CO on Pt and Pt-Ru[J]. Journal of the Electrochemical Society, 1999, 146: 1093-1098.
[46] ANDERSON M S. Locally enhanced Raman spectroscopy with an atomic force microscope[J]. Applied Physics Letters, 2000, 76: 3130-3132.
[47] LI Y, LI Y, PEI A, et al. Atomic structure of sensitive battery materials and interfaces revealed by cryo-electron microscopy[J]. Science, 2017, 358: 506-510.
Differential electrochemical mass spectroscopy: A pivotal technology for investigating lithium-ion batteries
ZHAO Zhiwei1,2, PENG Zhangquan1,2
(1State Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, University Chinese Academy of Sciences, Changchun 130022, Jilin, China;2University of Science and Technology of China, Hefei 230026, Anhui, China)
Safety issues of lithium-ion batteries are becoming prominent with its application in electric vehicle and large-scale energy storage fields. Flammable gases evolved from electrode|electrolyte interface play an important role on the batteries’ safety characteristics. Fortunately, differential electrochemical mass spectrometry (DEMS) as a powerful technology provides exploitable strategies for understanding the gas-related parasitic reaction mechanisms. The review covers the history, basics, know-how, and applications in safety researches of DEMS. In addition, emerging opportunities, critical challenges, future directions and strategies are particularly highlighted.
differential electrochemical mass spectrometry; lithium-ion battery; safety
10.12028/j.issn.2095-4239.2018.0131
TM 911
A
2095-4239(2019)01-001-13
2018-07-27;
2018-08-24。
国家自然科学基金项目(21733012,21633008,21605136,91545129,21575135),中国科学院战略性先导科技专项项目(XDA09010 401),吉林省科技厅项目(20150623002TC,20160414034GH),国家重点研发计划项目(2016YFB0100100)。
赵志伟(1993—),男,硕士研究生,主要研究方向为电化学质谱在锂电池中的应用,E-mail:zwzhao@ciac.ac.cn;
彭章泉,研究员,主要研究方向为能源界面电化学(电极动力学,现场光/质谱电化学,电极反应的第一性原理计算)、金属-空气电池、锂离子电池,E-mail:zqpeng@ciac.ac.cn。
