27%氧氟·二氯吡悬浮剂高效液相色谱分析方法
2018-12-11唐丽霖高婷婷许艳秋高立明吴春先
李 婷,唐丽霖,高婷婷,许艳秋,高立明,吴春先
(四川省农药检定所,成都 610041)
二氯吡啶酸(clopyralid)化学名称为3,6-二氯吡啶-2-羧酸,它是美国陶氏益农公司开发的一种具有吡啶羧酸结构的合成激素类除草剂,经由植物的根和叶吸收,并在植物体内传导。二氯吡啶酸常用于油菜、玉米、草坪等防除一年生阔叶杂草和多年生阔叶杂草(菊科和豆科杂草)[1]。乙氧氟草醚(oxyfluorfen)化学名称2-氯-α,α,α-三氟对甲苯基-(3-乙氧基-4-硝基苯基)醚,它为触杀型除草剂,在有光的情况下发挥除草作用。其主要经由胚芽鞘、中胚轴进入植物体,少量经根部吸收。乙氧氟草醚适合在苗前和苗后早期施用,防除阔叶杂草、莎草及稗草,对多种以种子繁殖的杂草有良好的防除效果,且对多年生杂草有一定的抑制作用[2]。目前国内报道的二氯吡啶酸分析方法多采用高效液相色谱,乙氧氟草醚的分析方法有气相色谱内标法、高效液相色谱外标法[3-5]。采用高效液相色谱同时对复配制剂中二氯吡啶酸和乙氧氟草醚进行定性、定量分析的方法尚未见报道。本文建立了高效液相色谱内标法对二氯吡啶酸与乙氧氟草醚复配悬浮剂进行分析,为其质量控制提供了简便、准确、有效的分析方法。
1 实验部分
1.1 试剂与仪器
Waters 2695高效液相色谱仪,带2996二极管阵列检测器和Empower工作站;色谱柱:ODS C18不锈钢柱(250 mm×4.6 mm)。
乙腈(HPLC级)、磷酸(分析纯)、联苯(分析纯);二氯吡啶酸标样(98%)、乙氧氟草醚标样(99%)、27%氧氟·二氯吡悬浮剂(18%乙氧氟草醚+9%二氯吡啶酸),四川利尔作物科学有限公司。
1.2 色谱操作条件
流动相:乙腈+0.1%磷酸水溶液,采用梯度洗脱,条件见表1。流速:1.0 mL/min;柱温:30℃;检测波长:235 nm;进样体积:3 μL。保留时间:二氯吡啶酸约为5.4 min,内标物联苯约为14.2 min,乙氧氟草醚约为16.3 min。

表1 梯度洗脱条件
上述液相色谱操作条件系典型操作参数,实际操作中可根据不同仪器特点,对给定的操作参数进行适当调整,以期获得最佳效果。典型的标样及27%氧氟·二氯吡悬浮剂样品高效液相色谱图见图1和图2。
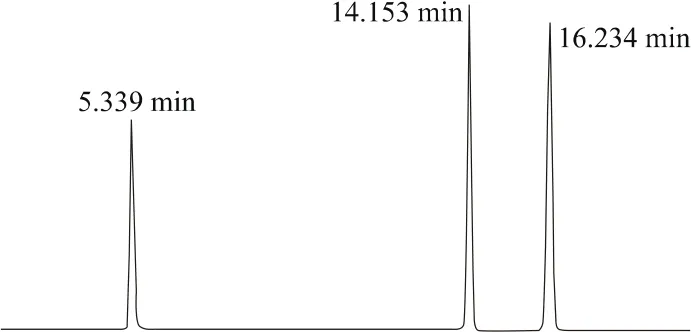
图1 标样液相色谱图

图2 27%氧氟·二氯吡悬浮剂液相色谱图
1.3 测定步骤
1.3.1 内标溶液的配制
称取约2 g(准确至0.000 2 g)联苯于500 mL容量瓶中,用乙腈溶解并定容,摇匀作为内标溶液备用。
1.3.2 标样溶液的配制
称取二氯吡啶酸标样约0.04 g(准确至0.000 2 g)、乙氧氟草醚标样0.08 g(准确至0.000 2 g)于50

式中:A1为标样溶液中二氯吡啶酸(或乙氧氟草醚)与内标物峰面积之比的平均值;A2为试样溶液中二氯吡啶酸(或乙氧氟草醚)与内标物峰面积之比的平均值;m1为二氯吡啶酸(或乙氧氟草醚)标样的质量,g;m2为试样的质量,g;P为标样中,二氯吡啶酸(乙氧氟草醚)的质量分数,%。mL容量瓶中,准确加入5.0 mL内标溶液,再用乙腈溶解并定容,摇匀,超声波溶解5 min,静置,备用。
1.3.3 试样溶液的配制
称取27%氧氟·二氯吡悬浮剂样品约0.45 g(准确至0.000 2 g)于50 mL容量瓶中,加入少量水溶解样品,准确加入5.0 mL内标溶液,用乙腈溶解并定容,摇匀,超声波溶解5 min,静置,备用。
1.3.4 测定
在上述色谱条件下,待仪器基线稳定后连续注入数针标样溶液,直至相邻2针中二氯吡啶酸(乙氧氟草醚)的响应值相对变化<1.5%,按照标样溶液、试样溶液、试样溶液、标样溶液的顺序进行测定。
1.3.5 计算
将测得的2针试样溶液以及试样前后2针标样溶液中的二氯吡啶酸(或乙氧氟草醚)与内标物峰面积的比值分别进行平均,试样中二氯吡啶酸(乙氧氟草醚)的质量分数w(%)按下式计算。
2 结果与讨论
2.1 流动相的选择
分别以不同比例的甲醇+水、乙腈+水、甲醇+0.1%磷酸水溶液、乙腈+0.1%磷酸水溶液为流动相,对试样进行分离检测。实验结果表明,当以乙腈+0.1%磷酸水溶液为流动相时,二氯吡啶酸、乙氧氟草醚和联苯能得到有效分离。因二氯吡啶酸极性强,保留时间较短,而乙氧氟草醚极性较弱,保留时间较长,采用等度洗脱难以有效分离,故选择梯度洗脱进行样品分离。在选定的洗脱条件下,三者的分离效果较好,峰形对称,且保留时间适中。
2.2 内标溶液的选择
以联苯、对氯苯酚、癸二酸二丁酯等为内标物,在相同色谱条件下对二氯吡啶酸及乙氧氟草醚的出峰情况以及吸收波长进行比较。结果显示,以联苯为内标物最为合适。
2.3 吸收波长的选择
通过Waters 2996二极管阵列检测器的光谱数据采集功能,获得二氯吡啶酸、乙氧氟草醚和内标物联苯200~400 nm的吸收波长扫描图(图3)。二氯吡啶酸在194、224、281 nm有较好的吸收;乙氧氟草醚在194~240 nm有较好的吸收;联苯在200、248 nm有较好的吸收。三者在195 nm处均有最大吸收波长,但受杂质影响较大。二氯吡啶酸和乙氧氟草醚在225~240 nm均有较好吸收,且联苯在235 nm也有较大吸收。在235 nm波长下,基线平稳,杂质吸收较小,主峰和杂质峰分离效果较好。故确定235 nm为本分析方法的检测波长。
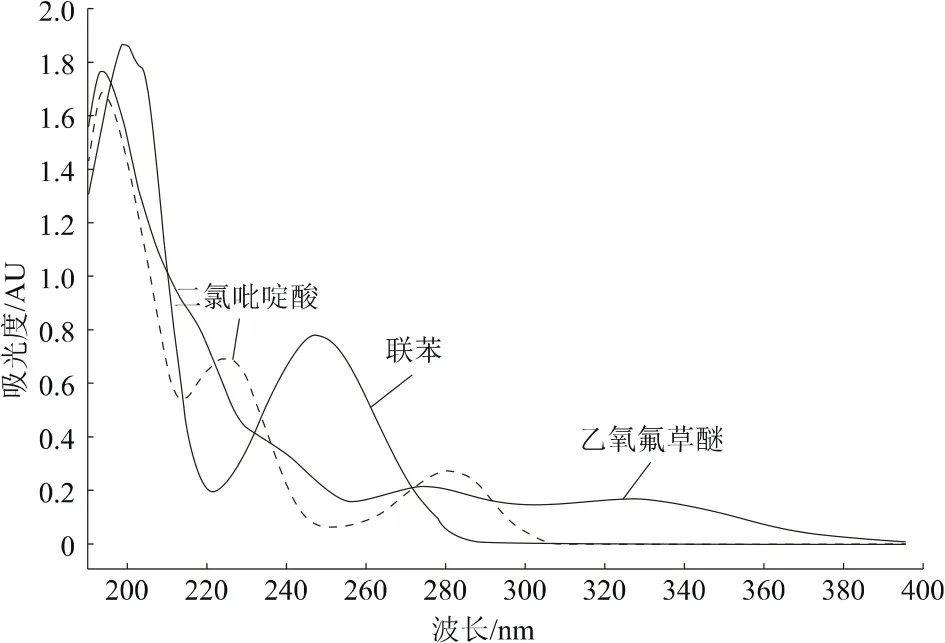
图3 二氯吡啶酸、乙氧氟草醚与联苯紫外吸收谱图
2.4 分析方法的线性相关性
分别称取二氯吡啶酸和乙氧氟草醚标样于50 mL容量瓶中,准确加入联苯内标溶液5.0 mL,用乙腈定容。分别配制5个不同质量浓度的标样溶液,在上述色谱操作条件下进行分析。以标样溶液质量浓度为横坐标,二氯吡啶酸(或乙氧氟草醚)与内标物联苯的峰面积之比为纵坐标,绘制标准曲线。二氯吡啶酸的线性方程为y=0.358 4 x-0.001 0,R2为1.000 0;乙氧氟草醚的线性方程为y=0.385 0 x-0.027 1,R2为0.999 3。
2.5 分析方法的精密度
从同一个27%氧氟·二氯吡悬浮剂中准确称取5个试样,在上述色谱操作条件下进行分析,实验结果见表2。
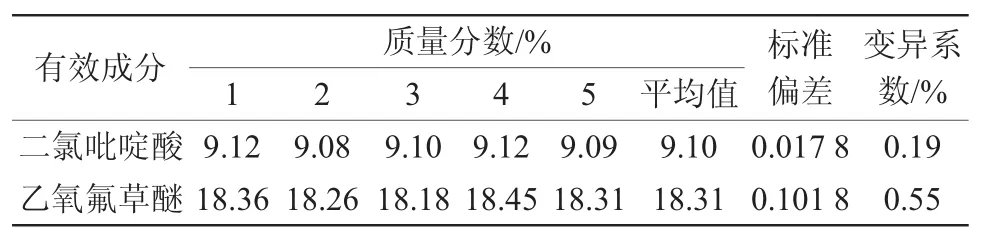
表2 分析方法的精密度实验结果
样品中二氯吡啶酸质量分数为9.10%,标准偏差为0.017 8,变异系数为0.19%;乙氧氟草醚质量分数为18.31%,标准偏差为0.101 8,变异系数为0.55%。
2.6 分析方法的准确度
以已知质量分数的27%氧氟·二氯吡悬浮剂样品为本底,分别加入一定量的二氯吡啶酸和乙氧氟草醚标样于50 mL的容量瓶中,准确加入5.0 mL内标溶液,用乙腈定容。按上述色谱操作条件测定二氯吡啶酸和乙氧氟草醚的质量分数。二氯吡啶酸的回收率为99.11%~101.75%,平均回收率为100.75%;乙氧氟草醚的回收率为100.30%~101.63%,平均回收率为101.03%,数据见表3。
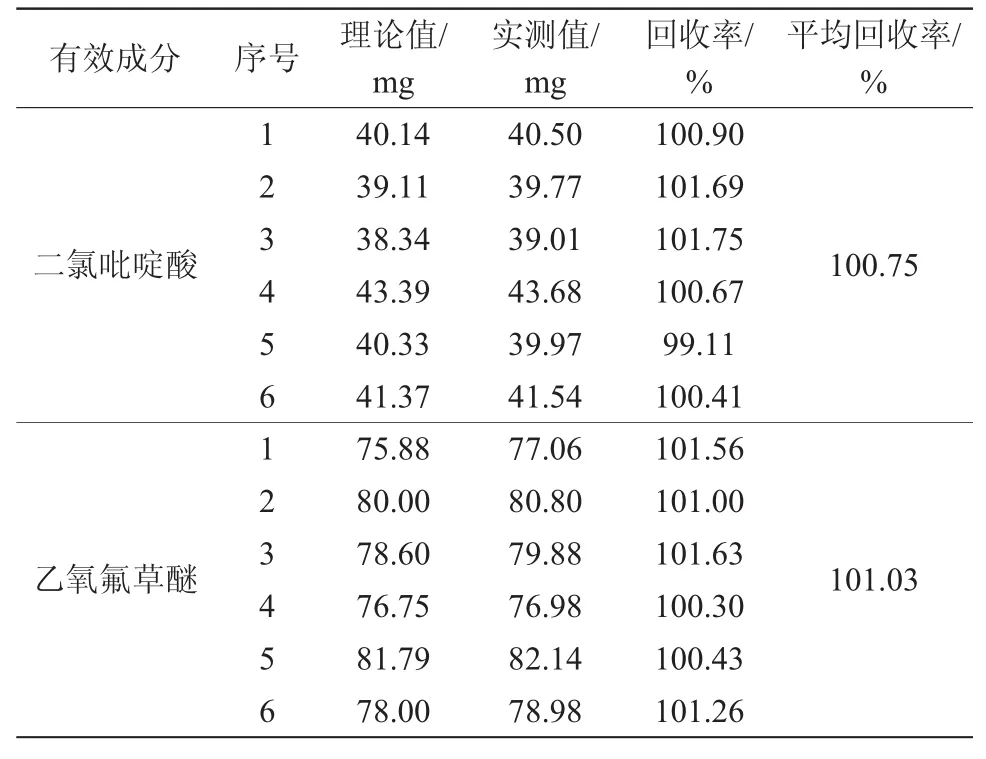
表3 分析方法的准确度实验结果
3 结论
本文通过对高效液相色谱分析条件的选择与优化,建立了27%氧氟·二氯吡悬浮剂的高效液相色谱内标分析方法。实验结果表明,本方法准确度和精密度高,线性关系良好,具有简便、快速的特点,是27%氧氟·二氯吡悬浮剂质量检测和监测可行的分析方法。
