磺胺嘧啶分子印迹电化学传感器的制备及其快速检测食品中磺胺嘧啶药物残留
2018-11-28赵玲钰秦思楠高文惠
赵玲钰,秦思楠,高 林,高文惠*
(河北科技大学生物科学与工程学院,河北省发酵工程技术研究中心,河北 石家庄 050000)
磺胺类药物是一类应用较普遍、效果良好的抗菌药物,主要对革兰氏阳性菌及一部分革兰氏阴性菌有较强的抑制作用,曾被广泛用于人类肠道和呼吸道疾病的治疗。长期以来,磺胺类药物在畜牧养殖业中起到较大作用,然而畜牧养殖业者对磺胺类药物的不科学使用及滥用造成一些动物源性食品中磺胺类药物严重超标[1-2]。经调查,畜禽养殖业中猪、牛及鸡的磺胺类药物使用量最大。因此,为保障人体健康,对该类畜禽肉中磺胺类药物残留的监管及检测不容忽视。我国对磺胺类及其增效剂的使用有比较明确的规定,GB 29694—2013《动物性食品中13 种磺胺类药物多残留的测定》中给出畜禽肉类产品13 种磺胺类药物的定量限为10 μg/kg[3];SN/T 4057—2014《出口动物源性食品中磺胺类药物残留量的测定》中规定出口类动物源性食品中磺胺类药物的测定低限为0.003 mg/kg[4];欧盟/470/2009中也明确规定,在所有动物源性食品中磺胺类药物残留总量不能超过100 μg/kg[5]。磺胺类药物中最具代表性,且使用量最大、使用效果较好的是磺胺嘧啶,其结构式如图1所示。
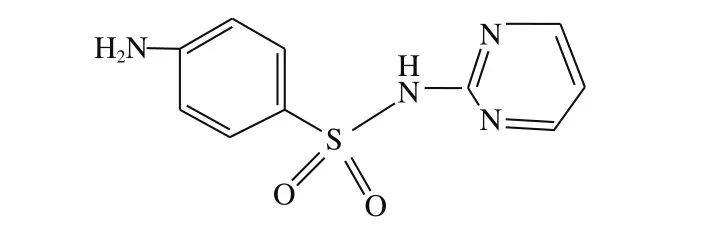
图1 磺胺嘧啶结构式Fig. 1 Structure of sulfadiazine
监控磺胺类药物残留最好的方法是建立一种操作简单、重复性好、灵敏度高的快速检测方法。目前,食品中磺胺嘧啶药物的检测方法大多采用高效液相色谱法[6-7]、色谱-质谱联用技术[8-12]以及免疫技术[13-14]等,然而,采用色谱法需要较为昂贵的分析仪器和专业技术人员,尤其存在样品前处理过程繁琐、有基质干扰、对低含量的目标物萃取效率低、样品检测成本高、难以实现快速检测等缺点;而免疫学方法需要制备抗体,制备药物抗体需要合成完全抗原并免疫动物,周期长,且生物活性受多种因素影响而易于失活。近年来,传感器法用于药物的检测以其检测快速、操作简单、价格低廉的特点越来越受到广大科研工作者的关注[15-18]。分子印迹电化学传感器将分子印迹聚合物选择性强、稳定性好的优势与电化学传感器特点相结合用于被分析物的检测可以得到良好的效果,如果同时将纳米材料例如纳米金、多壁碳纳米管等对电极进行修饰可以提高方法的灵敏度[19]。另外,模板分子和功能单体的选择、模板分子与功能单体比例的优化,对传感器的稳定性和选择性有着至关重要的作用,而紫外光谱法对于选择功能单体及优化模板分子与功能单体的比例起着重要作用。
目前,基于碳纳米管及纳米金溶胶(Au nano sol,AuNPs)修饰电极的磺胺类分子印迹电化学传感器的制备鲜有报道[20]。由于磺胺嘧啶电活性差,直接采用电化学分析法测定磺胺嘧啶灵敏度很低,因此本研究采用分子印迹技术借助电活性探针间接对动物源性食品中磺胺嘧啶或磺胺嘧啶钠残留进行测定,即以磺胺嘧啶为模板分子,邻氨基苯酚(o-aminophenol,OAP)为功能单体,引入紫外光谱法优化二者比例,用羧基化多壁碳纳米管(multi-walled carbon nanotube-COOH,CMWCNTs)和AuNPs以滴涂的方法修饰玻碳电极(glassy carbon electrode,GCE),电聚合形成OAP聚合膜,制备了磺胺嘧啶分子印迹电化学传感器,并将其应用于食品中磺胺嘧啶残留的快速检测。
1 材料与方法
1.1 材料、试剂与仪器
猪肉、牛奶、鸡蛋、鲜虾 市售。
磺胺嘧啶钠(纯度98%)、磺胺甲基嘧啶(纯度99.5%)、磺胺二甲基嘧啶(纯度98%)、OAP(纯度98%)、盐酸多巴胺(dopamine hydrochloride,DHY)(纯度98%)、壳聚糖(chitosan,CS)(脱乙酰度≥95%) 美国阿拉丁试剂有限公司;CMWCNTs德科岛金科技有限公司;氯金酸(含金量47.3%) 南京试剂厂;柠檬酸三钠、N,N-二甲基甲酰胺、铁氰化钾、亚铁氰化钾、氯化钠、磷酸二氢钠、磷酸氢二钠、醋酸、醋酸钠、高氯酸、高氯酸钠、乙腈、无水乙醇(均为分析纯);实验用水为超纯水。
LK98BII型电化学工作站 天津市兰立科化学电子高技术有限公司;三电极系统(CHI104 GCE(直径3 mm)、CHI115铂丝电极、CHI111 Ag/AgCl电极);抛光材料(氧化铝粉、麂皮) 上海辰华仪器有限公司。
1.2 方法
1.2.1 还原法制备纳米金胶体
纳米金的制备方法有多种,其中以水相中化学还原法最常用,化学还原法的原理是向一定浓度的氯金酸溶液中加入一定浓度还原剂,将金离子还原成金原子[21]。采用柠檬酸三钠在水相中还原氯金酸制备AuNPs,先将1 g氯金酸标准品一次性溶于水中,将溶液转移至100 mL棕色容量瓶中定容,配制成1%氯金酸溶液,再移至100 mL广口瓶放置阴暗处保存;在室温条件下参照Frens[22]的方法,在100 mL圆底烧瓶中加入50 mL水,向其中加入1%氯金酸溶液1 mL,120 r/min搅拌,并加热至微微沸腾后迅速加入1%柠檬酸三钠溶液2 mL,控制温度不变持续搅拌5 min,待溶液变为酒红色停止加热,再持续搅拌至溶液冷却至室温,将制备好的AuNPs取出,在60 ℃水浴条件下将液体蒸发至25 mL,得到的AuNPs浓缩液移至棕色瓶中冷藏保存。AuNPs最佳吸收波长为520 nm。
1.2.2 纳米金用量的优化
采用滴涂法将A u N P s修饰到电极表面。将CS-CMWCNTs复合物10 μL滴涂到已处理好的GCE表面,然后将浓缩后的AuNPs滴涂到修饰了CS-CMWCNTs复合物的电极表面,晾干。实验采用DPV法分别考察0、2、4、6、8、10、12 μL AuNPs对电极的修饰效果。
1.2.3 CS-CMWCNTs/AuNPs复合物的制备
将0.2 g CS溶解在20 mL 1%的醋酸溶液中,磁力搅拌至CS完全分散在醋酸溶液中且气泡全部消失,得到1%的CS溶液;将20 mg CMWCNTs粉末置于10 mL的N,N-二甲基甲酰胺中超声30 min,得到CMWCNTs分散液;再将1% CS溶液与CMWCNTs分散液等体积混合,磁力搅拌5 min后超声20 min得到CS-CMWCNTs复合物。取CS-CMWCNTs复合物10 μL及浓缩后所得AuNPs最佳剂量,二者混合得到CS-CMWCNTs/AuNPs复合物。
1.2.4 模板分子与功能单体的紫外光谱法测定
1.2.4.1 模板分子、功能单体及混合溶液紫外光谱扫描
分别配制1 mmol/L磺胺嘧啶钠溶液、6 mmol/L 2 种功能单体(OAP、DHY)溶液。取5 支10 mL比色管,分别向其中3 支比色管中加入磺胺嘧啶钠溶液200 μL、功能单体OAP溶液200 μL、DHY溶液200 μL;在另外2 支比色管中分别配制浓度比为1∶6的混合溶液:磺胺嘧啶钠溶液与邻苯二胺溶液各200 μL、磺胺嘧啶钠溶液与DHY溶液各200 μL。用水将5 支比色管定容至10 mL,超声10 min后,4 ℃冷藏静置14 h,然后进行紫外光谱扫描。
1.2.4.2 模板分子与不同比例功能单体的紫外光谱扫描
分别配制1 mmol/L磺胺嘧啶钠溶液、2 mmol/L 2 种功能单体(OAP、DHY)溶液。取10 支10 mL比色管,分成2 组,5 支为一组。分别向两组比色管中加入磺胺嘧啶钠溶液150 μL,按照模板分子与功能单体浓度比为1∶2、1∶4、1∶6、1∶8、1∶10向两组比色管中分别加入功能单体OAP溶液和DHY溶液150、300、450、600、750 μL。用水将10 支比色管定容至10 mL,超声10 min后,4 ℃冷藏静置14 h,对配制的混合液进行紫外光谱扫描。
1.2.5 GCE的预处理
将GCE依次用0.5、0.3、0.05 μm粒度的氧化铝粉在麂皮表面研磨抛光成镜面后再依次用无水乙醇、水超声清洗,氮气吹干,再将电极置于0.5 mol/L H2SO4溶液中,用循环伏安(cyclic voltammetry,CV)法扫描10 圈对电极进行活化处理,以达到电极最佳状态;取出电极再次用水冲洗后氮气吹干,然后将其置于含0.5 mol/L KCl和5 mmol/L K3[Fe(CN)6]-K4[Fe(CN)6]的溶液中采用CV扫描。重复以上操作直至得到对称且可逆的氧化还原峰,峰电位差在85 mV以下,最后取出电极待用[23]。
1.2.6 聚合条件的优化
实验选取醋酸-醋酸钠、磷酸盐缓冲溶液(phosphate buffer solution,PBS)、高氯酸-高氯酸钠3 种常用的聚合电解质溶液。分别考察功能单体OAP及磺胺嘧啶钠在醋酸-醋酸钠、PBS、高氯酸-高氯酸钠溶液及不同pH值条件下的溶解情况、聚合性质、成膜情况。在确定最佳电解质溶液和pH值后,根据功能单体的成膜电位分别选取了0~1.3、0~1.5、0~1.7、-0.2~1.3、-0.2~1.5、-0.2~1.7 V电位范围,采用差分脉冲伏安(differential pulse voltammetry,DPV)法和CV法对聚合效果进行考察。在其他条件相同的情况下,考察扫描圈数分别为5、10、15、20、25、30、40、50时,制备的聚合物膜对传感器性能的影响。
1.2.7 传感器的制备与洗脱
首先对预处理好的电极进行修饰,取1 0 μ L CS-CMWCNTs复合物分2 次(每次5 μL)滴涂到打磨干净的电极表面,随后用同样的方法将1.2.2节中选取的AuNPs最佳剂量滴涂到已修饰CS-CMWCNTs复合物的电极上,自然晾干,得到CS-CMWCNTs-AuNPs修饰电极;根据1.2.6节所得的最佳聚合条件,将上述电极置于聚合液中通过CV进行电化学聚合,在CS-CMWCNTs/AuNPs修饰电极表面得到含模板分子磺胺嘧啶的OAP电聚合膜;取另一支打磨好的GCE作为非印迹电极,处理方法除在电聚合过程中不添加磺胺嘧啶钠外,其余与上述方法完全一致。
聚合完成后,取出印迹电极,将电极分别置于甲醇-0.5 mol/L硫酸溶液(1∶4,V/V)、乙腈-0.5 mol/L硫酸溶液(1∶4,V/V)、甲醇-0.5 mol/L醋酸溶液(1∶4,V/V)中,对洗脱剂进行优化,在-0.2 V电位条件下进行电位诱导洗脱模板分子,自然晾干,得到磺胺嘧啶钠分子印迹电化学传感器。
1.2.8 电化学测量
实验过程中,在室温条件下用CV法和DPV法优化实验条件并表征电化学传感器性能,将三电极系统置于电活性探针溶液中测量电极的电流响应。测量参数如下:CV法,扫描电位和扫描速率分别为-0.2~0.8 V和50 mV/s;DPV法,起止电位为-0.2~0.6 V、电位增量0.004 V、脉冲宽度0.05 V、脉冲幅度0.05 V、脉冲间隔0.1 s。样品测量时,每次测量后,将工作电极浸泡在甲醇-0.5 mol/L H2SO4(1∶4,V/V)溶液中电诱导洗脱,除去模板分子后用水冲洗电极,氮气吹干,再进行下一次测量。实验采用计时电流法对模板分子进行电位诱导洗脱,测量参数如下:扫描电位-0.2 V,阶跃次数180,脉冲宽度2 s。
1.2.9 样品处理
取2 g捣碎和混匀的样品(猪肉、牛奶、鸡蛋、鲜虾)置于50 mL离心管中,加入5 mL乙腈提取剂,涡旋振荡混匀2 min,超声提取10 min,4 000 r/min离心10 min,取上清液,将剩余残渣加入5 mL乙腈重复提取1 次,合并2 次上清液,用乙腈定容至10 mL待用[24-25]。
1.3 数据统计
采用Origin 8.0软件对实验数据进行分析以及图形处理。
2 结果与分析
2.1 AuNPs修饰电极剂量的确定
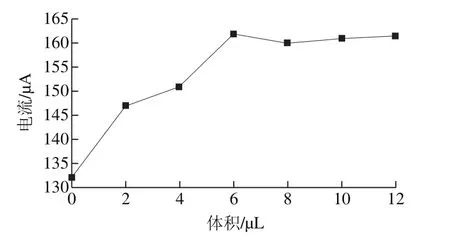
图2 AuNPs剂量的优化曲线Fig. 2 Optimization of nano-gold sol dose
如图2所示,在修饰剂量达到6 μL之前,峰电流值随修饰剂量的增加逐渐增大;当修饰剂量达到6 μL后,差分脉冲峰电流值不再增加,这是由于纳米金颗粒在电极表面分布达到饱和,再增加AuNPs的修饰剂量也不会再增大电极的比表面积和催化性能[26]。因此实验将AuNPs的修饰剂量确定为6 μL。
2.2 功能单体选择及功能单体与模板分子比例的确定
实验结合紫外光谱法对功能单体进行选择,并对模板分子与功能单体的作用机理进行研究。如果模板分子与功能单体不发生作用则同一波长条件下紫外吸光度应该是2 种物质吸光度的加和,即混合物的理论吸光度(E2);而模板分子与功能单体混合物的实际吸光度为该混合物在该波长条件下实测的紫外吸光度(E1);而且实际吸光度与理论吸光度的差值越大,表明二者的相互作用越大[27]。
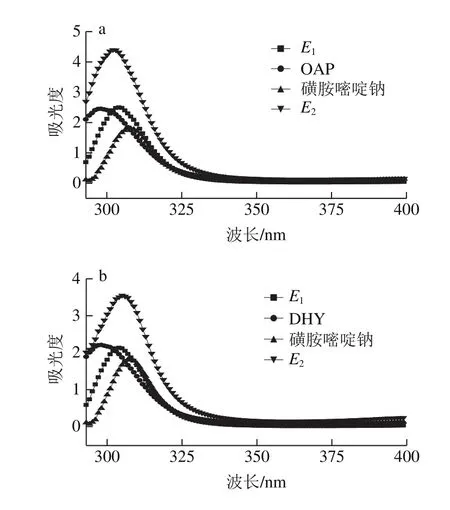
图3 模板分子、功能单体及其混合物紫外吸收光谱Fig. 3 UV absorption spectra of template molecule, functional monomer and their mixture
由于磺胺嘧啶难溶于水,而磺胺嘧啶与磺胺嘧啶钠主体结构相同,其与常用功能单体的作用形式和作用位点相同,故用磺胺嘧啶钠代替磺胺嘧啶进行相关实验。为了寻找合适的功能单体,根据模板分子磺胺嘧啶的结构和特性,考察2 种常用的功能单体OAP和DHY对磺胺嘧啶的作用效果。由图3可知,模板分子与2 种功能单体(OAP和DHY)的E2均高于E1,即理论吸光度大于实际测定值,说明模板分子与功能单体之间产生了相互作用。根据物质结构可以推断,这种作用为氢键作用力,2 种功能单体与模板分子的混合溶液的最大吸收峰出峰位置均发生了变化,最大吸收波长发生了红移。由图3还可知,磺胺嘧啶与OAP、磺胺嘧啶与DHY其E2与E1的差值分别为2.5与1.3,即磺胺嘧啶与OAP的E2与E1的差值较大,这意味着二者之间结合力较大,稳定性好,此为电聚合成膜提供了佐证和理论依据。因此选择OAP作为本实验功能单体。
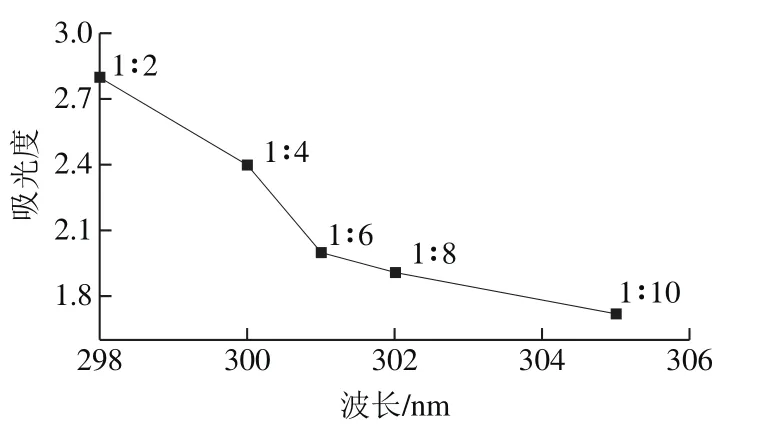
图4 不同比例的模板分子和OAP预组装体系最大吸收波长与吸光度变化趋势Fig. 4 Maximum absorption wavelength and absorbance from different ratios between template molecules and OAP
实验结合紫外光谱法确定模板分子与功能单体二者比例。如果模板分子与功能单体混合物体系最大吸收波长对应的吸光度变化趋于平缓时,说明混合物中模板分子与功能单体作用趋于稳定,此时平缓区的转折点所对应的混合物比例则拟定为二者的最佳比例[28]。如图4所示,磺胺嘧啶与OAP对应浓度比在1∶6~1∶10范围之间混合物体系最大吸收波长对应的吸光度波长变化趋于平缓。因此,可以推断磺胺嘧啶与OAP最佳浓度比为1∶6。
2.3 聚合电解质及其pH值的选择
检测所用电解质溶液的种类及pH值对响应信号的峰电流有显著影响。通过1.2.7节中的不同电解质对电极进行聚合,结果表明,当3 种聚合电解质溶液在弱酸性(pH 5.5)条件下,OAP均能在修饰的电极表面形成致密的不导电薄膜,适合用于制备分子印迹电化学传感器。实验随后采用DPV法考察以上聚合电解质溶液在相同聚合条件下聚合膜的响应性能;结果表明,当pH 5.5时,OAP在高氯酸-高氯酸钠电解质溶液中形成的聚合物膜最稳定、绝缘能力最好,并在模板分子洗脱后电流响应值最大,为113.3 μA,因此选择pH 5.5的高氯酸-高氯酸钠溶液为最佳聚合电解质溶液。
2.4 聚合扫描电位和扫描圈数的选择
由于聚合电位直接影响到修饰电极的洗脱效果,根据1.2.6节选取不同的电位对其进行考察。结果表明,当聚合电位为-0.2~1.7 V时模板分子洗脱较为困难,并且洗脱之后,氧化还原峰电流低;当聚合电位为-0.2~1.5 V时,峰电位正移,且氧化还原峰电流低;当聚合电位为-0.2~1.3 V时氧化还原峰电流值最大,且对称良好,因此实验选择-0.2~1.3 V为最佳电位。

图5 聚合圈数对峰电流的影响Fig. 5 Effect of polymerization cycles on the peak current
采用DPV表征,结果如图5所示,当扫描圈数为5~20 时,峰电流值随扫描圈数的增加而减小,这是由于聚合圈数较少时,膜太薄,OAP聚合膜在洗脱过程中已被破坏,GCE表面全部暴露,峰电流较大,如果扫描圈数增大,则聚合膜厚增加,膜稳定性增强,峰电流减小;随着扫描圈数的继续增加,在扫描30 圈时峰电流值达到最大,此时膜的稳定性处于最佳状态,且洗脱后印迹位点数量最多;然而,扫描圈数继续增加,峰电流值开始减小,这是因为聚合圈数增加使OAP聚合膜结构更致密更稳定,但膜厚度过大会造成模板分子无法从聚合膜中洗脱,印迹空穴减少,表征峰电流值减小,影响到传感器的灵敏度,因此实验选择扫描圈数30 圈为最佳。
2.5 分子印迹聚合物膜的制备

图6 OAP电聚合CV曲线Fig. 6 Cyclic voltammograms of OAP electropolymerization
当聚合圈数为30 圈,聚合速率为50 mV/s时,磺胺嘧啶分子印迹电化学传感器的电聚合曲线,如图6所示。由图6a可知,OAP在修饰电极表面是一个不可逆的过程,扫描第1圈时,在电位为0.3 V时出现氧化峰,且峰电流值达到60 μA;当扫描第2圈时,峰电流值明显降低;随着圈数的增加,峰电流强度不断减小,在聚合的最后一圈时峰电流值不足5 μA。以上现象表明,在OAP聚合过程中,在修饰电极的表面生成了一层致密不导电的聚合物薄膜,导致聚合液中的OAP在电极表面的电子传递受到抑制,无法进一步氧化,故电流响应降低。与图6a相比,图6b未见显著差异,表明OAP的电聚合不会因为磺胺嘧啶或磺胺嘧啶钠的存在而受到干扰,进一步说明磺胺嘧啶模板分子在电位范围内对OAP的电聚合不产生影响。
2.6 电位诱导法洗脱模板分子条件的选择
在探索模板分子洗脱方法时首先考察采用传统的溶剂浸泡洗脱法去除模板分子[29-30]。磺胺嘧啶分子中含有氨基和S=O基团,可与OAP的羟基和氨基以氢键作用力结合,故要求用于洗脱模板分子的溶液既能够破坏二者之间形成的氢键,又不能破坏OAP聚合物膜结构。磺胺嘧啶钠易溶于水、乙酸、盐酸、硫酸等溶剂,且在酸性溶液中较稳定,因此实验选取乙酸、盐酸、硫酸分别与甲醇、乙腈等组成的一系列的复合溶液作为洗脱剂,虽然不断调整酸与醇的比例与浸泡洗脱时间,但洗脱效果均不理想,这表明在本实验中洗脱模板分子的方法不适宜采用常规的浸泡洗脱法。因此,尝试将电位诱导法用于聚合膜中模板分子的洗脱,即电聚合后向修饰电极施加一定的电位以改变OAP与磺胺嘧啶聚合物内部的微环境,进而降低二者之间的氢键结合力,从而使磺胺嘧啶更容易从OAP膜内溶出以达到洗脱模板分子的目的。根据1.2.7节,将印迹电极分别置于不同洗脱剂中进行电位诱导洗脱模板分子,结果如图7所示。印迹电极在甲醇-0.5 mol/L硫酸溶液(1∶4,V/V)中洗脱后的响应电流值最大,效果最好,故实验确定甲醇-0.5 mol/L硫酸溶液(1∶4,V/V)为最佳洗脱剂。

图7 在不同洗脱剂中电位诱导洗脱后印迹电极响应信号Fig. 7 Response currents of the imprinted electrode after potentialinduced elution in different solvents
在确定甲醇-0.5 mol/L硫酸溶液(1∶4,V/V)为最佳洗脱剂之后,实验对诱导洗脱电位及诱导洗脱时间进行研究。在实验过程中发现当给予一个正的诱导电位进行洗脱时,洗脱后印迹电极在电活性探针溶液中未出现响应电流信号,说明在正电位条件下不能改变印迹膜的微环境,该膜在正电位条件下是稳定的;当在负电位条件下进行诱导洗脱时,随着负电位值的增加,响应信号逐渐增大,但是如果电位过负则导致印迹膜被破坏至脱落,响应电流值趋于裸电极的信号值,因此最终确定在-0.2 V条件下进行诱导洗脱。随后对模板分子洗脱时间进行考察,如图8所示,在电位诱导洗脱6 min时印迹电极的响应电流信号达到最大值,之后不再增加,故确定6 min为最佳的洗脱时间。

图8 诱导洗脱时间优化曲线Fig. 8 Optimization curve of induction elution time
综上所述,最佳的电位诱导洗脱条件为在甲醇-0.5 mol/L硫酸溶液(1∶4,V/V)中,施加-0.2 V的电位,诱导洗脱6 min。
2.7 吸附时间的选择

图9 吸附时间优化曲线Fig. 9 Optimization of adsorption time
实验采用DPV法对电极洗脱后再吸附的时间进行优化,将模板分子洗脱后的电极置于一定浓度的磺胺嘧啶钠溶液中浸泡不同的时间后,在铁氰化钾-亚铁氰化钾溶液中表征。如图9所示,随着浸泡时间延长,DPV峰电流值逐渐下降,当浸泡时间达到8 min后,峰电流值不再变化,表明聚合物膜对磺胺嘧啶或磺胺嘧啶钠的吸附已达到饱和,因此最佳吸附时间为8 min。
2.8 电极修饰和印迹效应表征
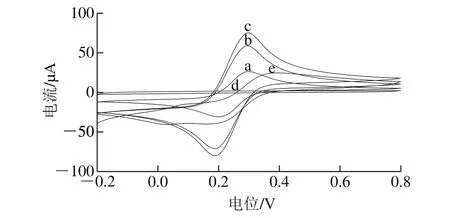
图10 电极在表征溶液中的CV曲线Fig. 10 Cyclic voltammograms of different electrodes in the probe solution
考察用滴涂法将CMWCNTs及AuNPs修饰到GCE的效果,并对不同电极进行表征,如图10所示。在铁氰化钾-亚铁氰化钾溶液中对裸电极进行CV扫描,其扫描曲线如图10曲线a所示,探针离子Fe3+/Fe2+在电极表面发生氧化还原反应,呈现一组氧化还原峰,峰电流值之比接近于1∶1;当在裸电极上修饰两层CMWCNTs后,有效的增大了电极的比表面积,并加快对电子的传递,从而使电极的响应峰电流明显增大,如图10曲线b所示;纳米金具有良好的生物相容性和良好的导电性能,CS-CMWNTs/GCE表面修饰AuNPs后的扫描结果如图10曲线c所示,响应峰电流又有了一定程度的增加。以上结果表明电极修饰效果良好。
实验对传感器的印迹效应进行了表征,由图10曲线d可以看出,聚合时不含磺胺嘧啶模板分子的非印迹电极在电诱导洗脱后的CV曲线为一条平滑的线,无氧化还原峰出现,表明当没有磺胺嘧啶存在时电聚合形成的OAP聚合膜结构稳定、紧密,不会因为电极施加-0.2 V电位而被破坏,探针离子无法到达电极的表面进行电子传递,所以无响应电流信号出现;而经过诱导洗脱模板分子后的印迹电极,其CV曲线出现了一组对称性稍差的氧化还原峰(图10曲线e),表明磺胺嘧啶存在时电聚合形成的OAP聚合膜电极在经过电位诱导洗脱模板分子之后留下一些带有特异性吸附的印迹空穴,使得探针离子可以到达电极表面发生电化学反应,因此有响应电流信号产生。以上现象表明磺胺嘧啶虽然不会影响到OAP的电聚合,但由于磺胺嘧啶与OAP之间以氢键结合,影响到了OAP膜的性质。
为了进一步验证磺胺嘧啶镶嵌到OAP聚合膜中,进行一组对比实验。将未经洗脱模板分子的印迹电极和洗脱模板分子后的印迹电极均置于pH 5.5的高氯酸-高氯酸钠溶液中进行CV扫描。对比实验结果表明,0.2~0.6 V范围内洗脱模板分子后的印迹电极出现氧化峰,未洗脱的没有出现氧化峰,出现的宽峰为嵌入膜内的磺胺嘧啶分子被洗脱后,探针离子发生了电化学氧化还原反应所致,峰电流较高且峰较宽,表明磺胺嘧啶与聚合物骨架存在分子间作用力,洗脱时模板分子与洗脱液发生反应,因此模板分子能从聚合物膜中较快地被洗脱到洗脱液中。
2.9 传感器选择性测定

图11 分子印迹电化学传感器的选择性Fig. 11 Selectivity of the molecularly imprinted electrochemical sensor
为考察分子印迹电化学传感器的选择性能,实验选取与磺胺嘧啶分子结构相似的磺胺二甲基嘧啶、磺胺甲噁唑以及与磺胺嘧啶分子结构相差较大的四环素作为干扰物质进行研究。将电极分别浸入到不同浓度(0.01、0.02、0.05、0.1、0.5 μmol/L)磺胺嘧啶钠溶液与干扰物质溶液中做吸附实验,采用DPV法考察电流响应,计算吸附目标物质前后相对峰值电流变化(ΔI)情况,ΔI越大,表明传感器对目标物质选择性越强。如图11所示,通过吸附前后响应电流的变化值(ΔI)可以看出,吸附相同浓度的磺胺嘧啶与干扰物质,印迹电极对模板物质的ΔI值最大,而对其他分子结构相似的物质响应较小,对与磺胺嘧啶结构相差较大的四环素几乎没有电流响应,表明该传感器抗干扰能力很强。
2.10 分子印迹电化学传感器的分析性能
2.10.1 分子印迹电化学传感器间接测定磺胺嘧啶的原理及测定结果
间接测定法是指以Fe3+/Fe2+为电活性探针,将洗脱模板分子后的电极置于探针溶液中用DPV法扫描,测定的峰电流值记为i0,然后向探针溶液中加入一定量的磺胺嘧啶钠,磺胺嘧啶游离嵌入到聚合物膜的空穴中,导致到达电极表面的探针离子减少,得失电子减少,峰电流下降,将此峰电流值记为i,将二者差值记为Δi=i0-i。磺胺嘧啶钠的浓度越大,同等时间内游离嵌入到聚合物膜空穴中的模板分子数量就越多,峰电流下降的越多,Δi越大。因此,根据Δi与磺胺嘧啶钠的浓度关系可以间接测定磺胺嘧啶或磺胺嘧啶钠。
2.10.2 线性关系与检出限测定结果

图12 不同浓度的磺胺嘧啶钠标准溶液DPV图Fig. 12 Differential pulse voltammograms of different concentrations of SD-Na standard solutions
配制一系列浓度(0.002~10 μmol/L)的磺胺嘧啶钠标准溶液,在最佳条件下,采用间接法研究磺胺嘧啶浓度与DPV法表征响应峰电流的关系。以响应峰电流差值(ΔI)为纵坐标,磺胺嘧啶钠标准溶液浓度的对数为横坐标,作图得出二者线性关系(图12),以3 倍信噪比确定目标物质的检出限。结果表明,磺胺嘧啶或磺胺嘧啶钠在1.0×10-8~2.0×10-6mol/L浓度范围内与ΔI呈线性关系,其线性方程为Y=-18.38 lgC-90,线性相关系数R为0.996 8,检出限为3.3×10-9mol/L,低于欧盟规定的可食用动物组织中磺胺嘧啶最低检出限量水平以及国标限定水平,因此本实验方法满足动物源性食品中磺胺嘧啶的检测要求。
2.10.3 回收率与精密度测定结果
在最佳实验条件下,样品(猪肉、牛奶、鸡蛋、鲜虾)在5 μg/kg和25 μg/kg 2 个添加水平条件下进行加标回收率和精密度实验,结果如表1所示。样品加标平均回收率在83.50%~97.80%之间,相对标准偏差(relative standard deviation,RSD)不大于4.0%。说明该方法的准确度和精密度良好。
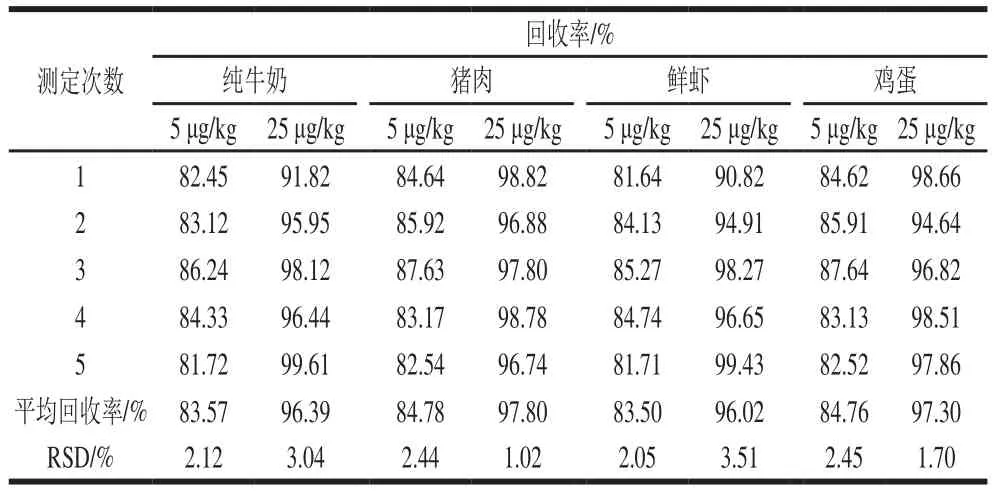
表1 样品加标回收率和精密度(n=5)Table 1 Recoveries and precision in spiked samples (n= 5)
2.10.4 重复性与稳定性结果
重复采用1.2.4节方法平行制备3 支印迹电极,分别对相同浓度的磺胺嘧啶钠标准溶液平行测定7 次,检测响应信号的RSD分别为1.42%、1.93%、2.11%,此结果表明电极的重复性良好。对该3 支电极连续使用20 次,检测响应信号分别降为最初值的86%、84%、89%,此结果表明印迹电极在有效使用次数内连续使用的稳定性良好。
2.10.5 实际样品的测定
取市售猪肉、牛奶、鸡蛋、鲜虾各5 个样品,按1.2.6节方法处理样品,取待测液各5 mL分别置于电解杯中,在1.2.5节电化学测量条件下进行测定。检测结果表明,被检样品均未检出磺胺嘧啶。
3 结 论
实验以磺胺类药物中常用的磺胺嘧啶为模板分子,选择OAP为功能单体,柠檬酸三钠还原氯金酸制备AuNPs与CMWCNTs修饰GCE,在修饰电极上成功制备了磺胺嘧啶分子印迹电化学传感器。采用CV法和DPV法研究了印迹传感器的电化学响应,优化了制备条件,考察了印迹传感器对模板分子以及结构类似物和其他干扰物质的选择性能,并将该传感器应用于实际样品的快速检测。结果表明,该传感器具有良好的印迹效应、灵敏度高、选择性强、稳定性好、检测准确快速,可用于动物源性食品中磺胺嘧啶药物的残留检测。
