一个深度学习DNA序列特异性的预测模型
2018-11-14黄立群丁雪松张步忠
黄立群,丁雪松,张步忠,吕 强,2
1(苏州大学 计算机科学与技术学院,江苏 苏州 215006 2(江苏省计算机信息处理技术重点实验室,江苏 苏州 215006)
1 引 言
DNA序列上存在一些特殊区域能够让特异性蛋白(转录因子)结合在该区域上,这就是DNA序列特异性.获知这种序列特异性对转录和选择性剪切这类基因调控过程有至关重要的作用.通常情况下,这种序列特异性是通过费时费力的生物实验获得.如今,像一些人类基因组库或者蛋白质库等数据样本变得越来越庞大,而且生物特征的维度也在不断的增加.这两点使得传统的生物实验手段受到极大的挑战,科学家们可能要花费大量的时间去挑选数据,做重复耗费资金的实验.但是大量的生物数据却能够给深度学习[1]模型提供充足的训练样本,验证样本,以及测试样本.近几年深度学习中的卷积神经网络[2](Convolutional Neural Network,CNN)由于其提取边缘特征的能力使其在图像视觉领域表现优异,递归神经网络[3](Recurrent Neural Network,RNN)在基于序列化问题的自然语言处理[4]方面取得了显著效果.那么递归神经网络在序列化的生物领域可能也存在很大的优势.
除去生物领域方法,目前预测DNA序列特异性的方法主要有以gkmSVM[5]为代表的传统机器学习和以DeepBind[6]为代表的深度学习这两大类方法,DeepBind和DeepSEA[7]是两款基于深度学习算法框架的软件.这两款软件将深度学习应用到特异性蛋白质序列结合问题上,并且在ENCODE[8]数据库上和传统的实验方法相比,DeepBind和DeepSEA表现得更好.2016年DK Gifford[9]等人利用卷积神经网络深度学习框架,在只有 DNA序列和标签的条件下进行了DNA和蛋白质的是否结合的实验,同样取得了很好的结果.
DeepBind是一种基于卷积神经网络的深度学习模型,该模型的输入是一条DNA序列,将输入的序列看作一张图,利用卷积层和池化层在输入序列上进行特征提取,再通过多层感知器网络对提取的特征进行运算输出,如果DNA能结合蛋白质就输出1,否则就输出0,类似图像领域中的二分类问题.
gkmSVM是一种基于支持向量机的分类打分模型,该模型可以用来检测DNA在细胞中的可及性,训练好的模型能够对任何DNA序列的可及性进行分类打分.gkmSVM方法首先通过输入的正样本集和负样本集计算出核矩阵,然后通过计算出的核矩阵以及两类样本数据训练模型,最后利用训练好的模型对DNA序列进行分类打分.LS-GKM[10]是gkmSVM在训练集容量上的一次升级,gkmSVM只能训练小于等于5000的样本数,当样本集大于5000条时软件会运行失败,LS-GKM解决了这一问题.
本文使用基于序列词向量的深度学习方法对DNA序列是否结合蛋白质进行预测,所有的实验数据来自ENCODE项目的690个测定实验使用的数据.每个实验数据包含若干条长度为101的DNA字母序列,并且每条DNA序列对应一个标签0或者1.如果DNA不能结合特异性蛋白就将标签设置为0,否则为1.将DNA序列看作自然语言处理中的一条句子序列,将DNA序列中的一些碱基的组合类比成单词,对蛋白质序列的处理就类似于自然语言对语句的处理.本文先用一种算法对DNA序列进行分词操作,然后利用词向量模型[11]对分词后的DNA序列训练生成序列词向量.将生成的序列词量作为本文深度学习模型的输入,再利用卷积神经网络提取序列中的高层特征,然后将卷积神经网络的输出作为双向LSTM[12]的输入,随后通过双向LSTM对特征进行运算累积输出,最后进行分类.我们把本文方法简称为Biovect_CNN_LSTM.本文将训练好的模型在ENCODE的690测定测试集上做了测试,并且和DeepBind方法和LS-GKM方法做了比较.
本文模型凭借序列词向量,以及双向LSTM在空间、时间上的优势,使得模型结果AUC的分布要优于LS-GKM和DeepBind方法.
2 Biovect_CNN_LSTM模型设计
2.1 序列词向量的预训练
传统方法利用one-hot方法对组成DNA序列的4种碱基A,C,G,T进行编码,这仅仅是单纯的将字母转成数字,而在生物领域不同碱基可以组合成具有生物特性的区域.本文将实验中的所有DNA序列按照生物领域中对序列分析经常使用的k-mer方法对DNA序列进行切分.在计算基因组学中,k-mer是指所有通过DNA序列测序读到的所有可能的子序列.这样一条长度为n的DNA序列经过k-mer算法切分后就会生成一条新的复杂序列,新序列以每k个碱基为一组一共n-k+1组的形式存在.k-mer算法过程就是利用一个游标从左往右滑动,每次选取k个碱基将这k个碱基存入新序列中,并将游标继续向右滑动,直至序列末尾.
k-mer通常被用作序列比对之前的第一步分析方法.特异性蛋白可以通过转录因子绑定到DNA特定区域,考虑到三个碱基编码一个氨基酸,因此本文的k选取3.
经过上述步骤将每条长度为101的DNA序列切分成3个碱基一词,一共99个词的新序列,每个序列词相当于由3个字母组的一个单词,99个序列词相当于99个单词组成的一条句子,这样每个序列词之间可能存在生物相关性.再利用词向量模型训练所有的处理后的序列语料库,生成序列词向量.文中使用的词向量模型是CBOW模型,根据上下文预测目标词出现的概率,每个目标词用16维的向量表示.经过若干轮的迭代,将原先每条长度为99的碱基组序列,训练生成(99,16)的张量数据.
2.2 模型结构描述
本文使用的深度网络模型如图2所示.深度网络模型的输入是N×T,T是每条DNA序列中含有的序列词向量的个数,本文实验每条序列的词向量个数是99,N是CBOW模型生成的序列词向量的维度.

图1 深度模型结构
图1中模型的输入是一个序列Xt,此处t是99,对应每条序列由99个序列词构成,每个X是通过上文方法提取的16维的序列词向量. 将序列词向量序列输入模型第一层的1D卷积层,利用p×m的卷积核去扫描输入的序列,其中p是卷积核的个数,m是一维卷积核的长度,每次对m个序列词向量进行卷积.该卷积层相当于一个特征扫描器,目的是希望能在生物词向量的基础上提取出隐含的高层特征,挖掘更多的生物特征.
网络的第二层与传统的卷积神经网络不同,此处利用批规范化层(Batch Normalization)[13]代替传统的池化层(pooling).使用批规范化层能避免在训练过程中落入饱和区域导致的梯度消失,加速网络训练,使得网络收敛速度加快.也能避免因为使用池化后丢失部分特征这一缺点.批规范化层将每个隐藏层的输出结果在batch上进行批规范化后再将结果输入下一层,就像我们在数据预处理中将χ进行规范化后送入网络的第一层一样.
第三层是一个双向的LSTM层,之所以使用双向LSTM是因为DNA是有两条链组成的双螺旋结构.双向LSTM每个神经元的输出是由当前神经元的输入和该神经元左右邻居神经元经过计算得到.最后将所有的特征累计到序列的最后一位,作为全连接层的输入.
本文还在全连接层之间加入了Dropout层.Dropout的作用是随机的屏蔽一定比例的神经元,这些被屏蔽的神经元不参与层的计算,这样可以有效的防止模型过拟合.最后的输出层只有一个神经元,对应分类的两种结果0和1,如果DNA序列能和蛋白质绑定那么模型就输出1,否则模型输出0.
2.3 训练模型
本文的深度学习模型利用keras框架搭建,在CentOS7环境下进行训练和测试,硬件资源使用的是两块K20GPU.通过两块K20并行来加速训练.
模型的输入是由生物词向量组成的DNA序列,模型的minibatch是1024条DNA序列.训练集包含15960000条DNA序列,验证集的DNA序列条数是1773469条,总的测试集DNA序列条数是5113218条,通过监测模型的val_loss来判断模型是否可以提前结束训练,本文当val_loss在6次迭代都没有一点下降时提前结束训练.模型训练时间超过100小时.
模型使用Adam[14]优化器指导模型训练,这样在训练过程中无须复杂的调参过程.且计算高效占用内存小.
3 结 果
3.1 实验数据和评价指标
本文的实验数据来自ENCODE项目的690个不同的测定实验使用的数据,每个实验数据都有各自的训练集和测试集,本文利用所有的训练集训练模型,并且将训练集中的10%的数据划分为验证集,然后将训练好的模型分别在690个测试集上进行测试.每条DNA序列都是由A,C,G,T四个字母组成的字符串,4种字母代表生物领域中的4种碱基.所有数据由正样本数据集和负样本数据集组成,标签为1是正样本,标签为0是负样本.这些数据是本文的方法和其他方法比较的基础.
二分类的指标很多,ACC、Sec、Pec、recall、MCC等,但是最能综合反映模型性能是AUC(Area Under Curve).指标.所以,本文在AUC指标上进行了总体比较,并在几个典型的数据集上进行了个案比较.
(1)
M是正类样本的数目,N是负类样本的数目,通过对样本score从大到小排序,最小score对应样本的rank1,以此类推ranki.
3.2 总体性能分析
统计690个测试数据集上的AUC分布来评价本文模型的好坏,并且和DeepBind,LS-GKM的结果做了比较,结果如图2所示.
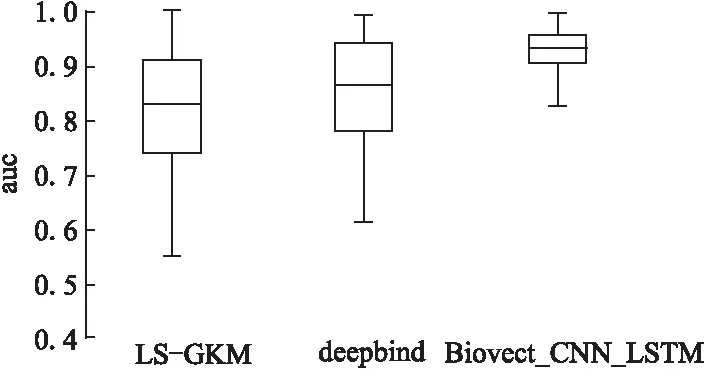
图2 三种方法的AUC分布
图2是三种方法的总体比较,纵坐标是AUC,横坐标是三种比较的方法.从图中可以看出LS-GKM方法的AUC结果主体分布在0.74~0.91之间,DeepBind方法的AUC主体分布在0.78~0.94之间,而本文模型的AUC主体分布在0.89~0.95之间.三个模型的AUC最高都接近1.这说明本文的模型训练结果要优于另外两个模型.
3.3 部分转录组数据集对比
本文从690个ENCODE测定实验数据中随机挑选5个实验数据结果进行比较,这5个实验集都是DNA绑定蛋白中的转录实验,转录是以DNA中的一条单链为模板,游离碱基为原料,在DNA依赖的RNA聚合酶催化下合成RNA链的过程.转录是蛋白质合成的第一步,对研究遗传有着起着举足轻重的作用,结果如图3所示,图3是三种方法在AUC指标上的性能比较,数据集描述见表1.在测试集4上DeepBind的AUC小于0.85,LS-GKM小于0.9,本文的方法已经接近0.95,

图3 5个实验数据集的结果
测试集5上DeepBind方法的AUC略低于0.9,LS-GKM的AUC略高于0.9,本文的方法依然接近0.95,其余三个样本集上三者方法AUC差距不大都接近1,由此可见本文的方法优于其他两种方法,尤其在测试集4,5上,本文的方法有着巨大的优势.

表1 5个实验数据集合名表
3.4 小结
上述实验结果表明,本文的基于序列词向量的深度学习模型在预测DNA和蛋白质结合要优于DeepBind和LS-GKM.原因在于本文的深度学习模型中,用一层双向LSTM层,LSTM通过门的机制解决梯度消失问题,而且LSTM能够捕捉长距离碱基的相互作用.
在DNA和蛋白质结合问题上,本文提出一种新的深度学习模型.实验结果表明,该方法的性能要好于目前的LS-GKM和DeepBind两种方法.
未来进一步研究可以着手于以更大规模的全基因序列库训练更加准确的序列词向量.另外在深度学习模型的设计中,尝试更加简化模型结构,提升模型的可解释性;可视化并解释模型提取的中间层特征等.
