Comparative transcriptome study provides insights into acquisition of embryogenic ability in upland cotton during somatic embryogenesis
2018-11-10SUNRuibinTIANRuipingMADanWANGShaohuiandLIUChuanliang
SUN Ruibin,TIAN Ruiping,MA Dan,WANG Shaohuiand LIU Chuanliang,2*
Abstract Background:The conversion from non-embryogenic callus(NEC)to embryogenic callus(EC)is the key bottleneck step in regeneration of upland cotton(Gossypium hirsutum),and hinders the transgenic breeding of upland cotton.To investigate molecular mechanisms underlying acquisition of embryogenic potential during this process,comparation analysis of transcriptome dynamics between two upland cotton cultivars with different somatic embryogenesis abilities was conducted.Results:Differentially expressed genes involved in the transformation from NEC to EC were detected in the two different cultivars.Principal component analysis based on DEGs showed that the NEC tissues of the two cultivars were highly heterogeneous,whereas the derived EC tissues were similar,which suggested the homogeneousness of EC between different lines.In the highly embryogenic cultivar CCRI 24,more of these genes were down-regulated,whereas,in the recalcitrant cultivar CCRI 12,more were up-regulated.Bioinformatics analysis on these DEGs showed that the vast majority of differentially expressed genes were enriched in metabolism and secondary metabolites biosynthesis pathways.Flavonoid biosynthesis and phenylpropanoid biosynthesis pathways were enriched in both cultivars,and the associated genes were down-regulated more in CCRI 24 than in CCRI 12.We deduced that vigorous secondary metabolism in CCRI 12 may hinder primary metabolism,resulting in tardiness of cell differentiation.Interestingly,genes involved in the plant hormone signal transduction pathway were enriched in the recalcitrant cultivar CCRI 12,but not in CCRI 24,suggesting more radical regulation of hormone signal transduction in the recalcitrant cultivar.Signal transduction rather than biosynthesis of plant hormones is more likely to be the determining factor triggering NEC to EC transition in recalcitrant cotton lines.Transcription factor encoding genes showed differential regulation between two cultivars.Conclusions:Our study provides valuable information about the molecular mechanism of conversion from NEC to EC in cotton and allows for identification of novel genes involved.By comparing transcriptome changes in transformation from NEC to EC between the two cultivars,we identified 46 transcripts that may contribute to initiating embryogenic shift.
Keywords:Upland cotton,Transcriptome,Non-embryogenic callus(NEC),Embryogenic callus(EC),Somatic embryogenesis
Background
Upland cotton(Gossypium hirsutum)is an important economic crop,grown in many countries,as the major natural fiber,an important raw material for textile,and nutrient-rich edible oil that can be extracted from cotton seeds(Sunilkumar et al.2006;Shi et al.2006).For many years,many efforts have been made to improve cotton yield,fiber quality,stress tolerance,and other traits.In recent decades,genetic modification technologies have been widely applied in the breeding of many plants,including cotton.Incorporating exogenous genes into explants by Agrobacterium-mediation or gene gun bombardment,and regeneration of transformed cells through somatic embryogenesis(SE)is the main and the most effective method used to produce transgenic cotton(Liu et al.2014;Zhang et al.2000;Leelavathi et al.2004).Genetic modification technologies have great potential to improve desirable cotton traits,including yield,fiber quality,and pest,disease,drought,and herbicide resistance.However,cotton genetic modification is severely constrained by the poor regenerative capacity of cotton plants(Zhang et al.2011;Shang et al.2009).Many factors,such as the explants,culture medium components,and light and temperature during culture,affect the regeneration of cotton cells(Wu et al.2004;Chi et al.2004).Nevertheless,the inherent genetic background is the essential limiting factor(Trolinder and Chen 1989;Dong 1991;Deo et al.2010).Although lots of effort have been made to optimize the somatic regeneration culture system,only very few upland cotton cultivars can be regenerated from Agrobacterium-mediated or biolistic genetic transformed cells(Leelavathi et al.2004;Shang et al.2009;Zhang et al.2000;Rajeswari et al.2010;Xie et al.2007;Zhang et al.2009;Sun et al.2009;Dong 1991;Liu et al.2004;Han et al.2009).
Plant regeneration through SE is a long and complicated process.First,the transformed cells dedifferentiate into calli on the culture medium.After several weeks,an embryogenic callus(EC)is acquired and then differentiates into an embryo,which ultimately grows into a plant.The process of converting a non-embryogenic callus(NEC)into an EC is the key bottleneck step(Zhang et al.2011;Shang et al.2009;Wu et al.2004;Rajeswari et al.2010).SE is a complex dynamic process that involves many intracellular metabolic changes and is influenced by various external environment factors.The underlying temporal-specific expression of genes plays an important role in this process.Analyzing the transcriptional activity of genes during the transition from NEC to EC may help to reveal the molecular mechanisms involved in the acquirement of embryogenic potential,which,in turn,may provide ideas to facilitate the induction of EC to further promote regeneration of a wider range of cotton cultivars for genetic modification.Differential-display of reverse transcription polymerase chain reaction(PCR),cDNA-amplified fragment length polymorphisms,suppression subtractive hybridization,and microarrays have been used to study gene expression during cotton SE,and a number of specifically or differentially expressed genes(DEGs)associated with cotton SE have been identified(Wu et al.2009;Zeng et al.2006;Zhu et al.2008;Leng et al.2007),but the data were insufficient or incomplete.Next-generation sequencing combined with bioinformatics analyses have proved to be powerful tools for more comprehensive genome and transcriptome analysis.Using RNA sequencing-based digital gene expression technology,Yang et al.(2012)reported the first genome-wide gene expression profiling of different SE stages(from hypocotyl to cotyledon embryos)in cotton.A total of 3 329 genes linked to various pathways were found to be differential expression during the transition from NEC to EC,and 137 genes were specifically switched on or off in this process.Xu et al.(2013)performed RNA sequencing to study the transcriptome changes in two upland cotton lines with different somatic embryogenic capacities in response to SE induction.Focusing on transcripts associated with phytohormone metabolism,seven auxin-related transcripts and nine cytokinin-related transcripts were specifically found in embryonic calli of a highly embryogenic line.All of these existing transcriptome studies on cotton SE by RNA sequencing are based on a de novo assembled transcriptome,which may contain many mistakes as the high complexity of plant genome.Now that genome sequencing of upland cotton has been completed(Li et al.2015),transcriptome sequences can be obtained more easily and accurately,making it conducive for transcriptome analysis.Here we conducted a comparative analysis on transcriptomes dynamics during NEC to EC conversion process between a recalcitrant embryogenic cotton cultivar and a highly embryogenic cotton cultivar by RNA sequencing.Different from previous studies,we mapped sequencing reads to the published genome as reference to assemble transcriptome.Besides,we also obtain EC from a recalcitrant cultivar,that the transcriptome difference between NEC and EC was compared,rather than just compare the difference of NEC before inductive treatment with after treatment.
Methods
Plant materials and induction of callus
CCRI 12 and CCRI 24,two commercial planting varieties of upland cotton in China,were used in our study.CCRI 12 is an excellent cultivar that has superb disease resistance and high yield,and produces high quality fiber.Because of its superior comprehensive performance,CCRI 12 has large acreage,and is an important material in cotton breeding.However,CCRI 12 shows recalcitrantembryogenesisfrom explantin genetic transformation,which hinders its application in transgenic breeding.CCRI 24 is the other major cultivar with high embryogenesis efficiency in China,and it has been developed into a model cultivar for cotton genetic transformation.We clarified the embryogenic differences between CCRI 12 and CCRI 24 by a regeneration test as described below.
Seeds were delinted using concentrated sulfuric acid,then sterilized in 3%hydrogen peroxide solution for 12 h,and rinsed with sterile water for five times before sowing on solid medium containing Murashige&Skoog(MS)(Murashige and Skoog 1962)macroelements salts,sucrose,and agar.After culturing at 28°C under a 16 h light/8 h dark photoperiod,the hypocotyls of 7-day-old seedlings were cut into 1 cm long segments,then placed on calli induction medium containing MS,0.1 mg·L-13-indole acetic acid(IAA),0.1 mg·L-1kinetin(KT),0.1 mg·L-12,4-dichlorophenoxyacetic acid(2,4-D),30 g·L-1glucose,and 2.5 g·L-1Gelrite,adjusted to pH 6.0.NEC of each cultivar were sampled from three different seedlings as biological replicates after 40-day culture under the same conditions as seedling growth,the number of NEC was counted before sampling.Then,the calli were transferred to subculture medium,in which all the exogenous hormone levels were reduced to 1/10 of those in the calli induction medium.EC was sampled from three different seedlings as biological replicates when the EC of each cultivar formed finally,after subculturing several times.The number of EC was counted before sampling.All the calli tissues were frozen in liquid nitrogen and stored at-80°C for RNA extraction.The NEC and EC of CCRI 12 and CCRI 24 represented four different sample types-named,respectively,by AN(NEC of CCRI 12),AE(EC of CCRI 12),BN(NEC of CCRI 24),and BE(EC of CCRI 24),with each sample type containing three samples from different individuals as biological replicates.These 12 samples were used forsubsequentRNA libraryconstruction and sequencing.
We quantified the embryogenic ability by computing the regeneration rate of NEC(the ratio of NEC to explant),and the regeneration rate of EC(the ratio of EC to NEC).
RNA extraction
Total RNA was extracted using a TIANGEN RNAprep Pure Plant Kit,following the manufacturer’s protocol(Tiangen n.d.).The quality of total RNA was tested using a NanoDrop 2000 instrument(Thermo Scientific)and agarose gel electrophoresis.The RNA samples were used for library construction and sequencing.
Library construction and RNA sequencing
We provided 20 μg of total RNA to the Beijing Genomics Institute(BGI)where the libraries were constructed and sequenced on an Illumina HiSeq 2000 platform.Briefly,mRNA was enriched using oligo-dT magnetic beads and fragmented into short fragments.cDNA was synthesized using the mRNA fragments as templates primed by random hexamers.Then,the cDNA fragments were purified for end repairing and single nucleotide A (adenine)addition,then connected with adapters.Fragments of about 250 base pair(bp)were selected by agarose gel electrophoresis,and purified as templates for PCR amplification.After PCR,qualification and quantification of the libraries were conducted using an Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-Time PCR System.The qualified libraries were sequenced using an Illumina HiSeq™2000 sequencer.
Genome mapping and bioinformatics analysis of(DEGs)
The raw sequence data were filtered to remove adapter sequences,reads with>10%of unknown bases,or>50%of low quality bases.The filtered clean data were used for further analysis.The BWA(Li and Durbin 2009)and Bowtie2(Langmead and Salzberg 2012)software were used to map the clean reads to the published Gossypium hirsutum TM-1 genome(Li et al.2015)and gene reference sequences,respectively.The mapping information was used to detect specific and common genes between the different samples.The expression levels of the identified genes and isoforms were quantified using the RSEM(RNA sequencing by Expectation Maximization)(Li and Dewey 2011)software package.Fragments per kilobase of transcript per million mapped reads(FPKM)values were calculated to evaluate expression levels of genes or transcripts.Based on the results,correlation and principal component analyses were conducted pair-wise between samples.The NOIseq software package(Tarazona et al.2012)was used to screen differential expression analysis based on the noise distribution model,genes with fold change ≥2,and divergence probability ≥0.8 were recognized as DEGs.Genes with similar expression patterns usually have some functional correlation.A hierarchical clustering analysis was performed of DEGs among the sample pairs using Cluster and Java Treeview software(Saldanha 2004).The Gene Ontology(GO)(Ashburner et al.2000)database and canonical pathways in the Kyoto Encyclopedia of Genes and Genomes(KEGG)(Kanehisa and Goto 2000)and plant gene datasets selected were used for GO functional and pathway enrichment analyses of the DEGs.
qRT-PCR validation
Quantitative real-time PCR(qRT-PCR)was carried out to verify the expression profile results calculated from the RNA sequencing data.We selected 13 genes that were differentially expressed during SE for qRT-PCR validation.The PCR primers were designed using the NCBI online Primer-BLAST.RNA samples were treated to remove genomic DNA and reversely transcribed using a TransGen AH341 kit.The obtained cDNA templates were diluted five times prior to amplification.The qRT-PCR reaction system contained 1 μL cDNA templates,0.5 μL each of 10 μmol·L-1forward and reverse primers,2×TransStart Top Green qPCR Super Mix,add ddH2O up to 20 μL.All the reactions had three technical replicates and were run on a Roche Light Cycler 480 instrument.The amplification program was as follows:pre-incubation at 95°C for 5 min,followed by 40 cycles of denaturation at 95°C for 10 s,annealing at 60 °C for 10 s and extension at 72 °C for 15 s.A melting curve was constructed to detect the specificity of the products.GhHis3(GenBank:AF024716)was used as the reference gene.The relative expression values were computed by the 2-ΔCTmethod.
Results
Obtaining and characterizing the NEC and EC tissues of CCRI 12 and CCRI 24
After 40 days culture on callus induction medium,both CCRI 12 and CCRI 24 produced NEC tissues.The calli of CCRI 12 were firmer,smoother,and faster growing than those of CCRI 24.After subculturing three times,the CCRI 24 calli began forming EC tissues,whereas the CCRI 12 calli remained non-embryogenic.CCRI 12 needed more times of subculturing before finally forming EC tissues.There was no obvious morphological differences between the EC tissues of CCRI 12 and CCRI 24,all of which were faint yellow,and had loose texture and a granular surface(Fig.1).
The regeneration rate of NEC showed no significant differences between CCRI 12 and CCRI 24,whereas the induction emergence rate of EC from NEC was notably different.The regeneration rate of EC in CCRI 24(91.9%)was five times that in CCRI 12(17.6%),suggesting that the differenceofembryogenesisbetween CCRI12and CCRI 24 was mainly due to the efficiency of induction from NEC to EC(Table 1).
Summary of the RNA sequencing data
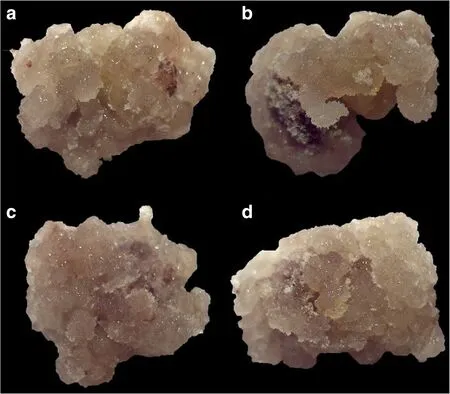
Fig.1 Non-embryogenic callus(NEC)and embryogenic callus(EC)tissues morphology of CCRI 12 and CCRI 24.NEC were induced from hypocotyl segment after 40 days culture on calli induction medium.a NEC of recalcitrant cultivar CCRI 12,represented as AN.b NEC of highly embryogenic cultivar CCRI 24,represented as BN.NEC were subcultured on medium with 1/10 of hormones to generate EC.c EC of recalcitrant cultivar CCRI 12(after subculturing for five times),represented as AE.d EC of highly embryogenic cultivar CCRI 24(after subculturing for three times),represented as BE

Table 1 Regeneration rates in different stages of SE in CCRI 12 and CCRI 24
After filtering the raw data,we obtained a total of 598.2 million clean reads of 90 bp in length for further analysis.Each library produced at least 4.37 G bases clean data,which were sufficient for gene expression profiling.Samples which showed bad correlations with other duplicates were removed.An average of 79.5%clean reads were mapped to the reference genome,and an average of 71.3%clean reads were mapped within known genes.Integrating sequencing data of twelve samples,a total of 60 908 genes(79%of the annotated genes in the reference genome)were detected.80.7%of these genes were common in the four libraries.1 121,850,803,1 136 genes were specifically expressed in AE,AN,BE,and BN,respectively.
qRT-PCR validation of gene expression from the RNA sequencing data
We selected 13 genes for qRT-PCR to validate gene expression levels obtained by our analysis of RNA sequencing data.SPSS v.22 wasused to calculate the correlation coefficient between qRT-PCR results(represented as 2-ΔCT)and the results derived from the RNA sequencing data(represented as FPKM value).The 2-ΔCTand FPKM values were both logarithmic transformed beforehand.The results showed high correlation between the expression levels obtained from the RNA sequencing data and qRT-PCR(Fig.2),which validated the results of the RNA sequencing expression profile analysis.
Principal component analysis(PCA)shows homogeneousness of EC between different cultivars
Based on the gene expression data,PCA clustering showed that NEC samples of CCRI 12(AN)separated clearly from NEC samples of CCRI 24(BN).On the contrary,EC samples of CCRI 12(AE)and CCRI 24(BE)clustered together,suggesting homogeneousness of EC tissues between CCRI 12 and CCRI 24(Fig.3).
Global analysis of differential gene expression during the transformation from NEC to EC
We detect DEGs between different callus states and different cultivars.The AN vs AE and BN vs BE comparisons were set to detect gene expression changes between NEC and EC tissues in CCRI 12 and CCRI 24,respectively.The AN vs BN comparison was set to detect gene expression differences between NEC tissues of CCRI 12 and CCRI 24,and the AE vs BE comparison was set to detecte gene expression differences between EC tissues of CCRI 12 and CCRI 24.A total of 3 720 DGEs were obtained in the four comparison pairs and used for further analysis.Statistics of DEGs number detected in four comparisons pairs is shown in Fig.4a.During the transformation from NEC to EC,2 779 and 2 364 DEGs were detected in CCRI 12 and CCRI 24,respectively,1 594 were detected in both cultivars.In CCRI 12,more DEGs were up-regulated than down-regulated,whereas,in CCRI24,morewere down-regulated than up-regulated(Fig.4a).When comparing different cultivars,490 DEGs were detected in NEC tissues between CCRI 12 and CCRI 24,while only 167 DEGs were detected in EC tissues between them.Figure 4b shows the overlap degree of DEGs detected in four comparing pairs.
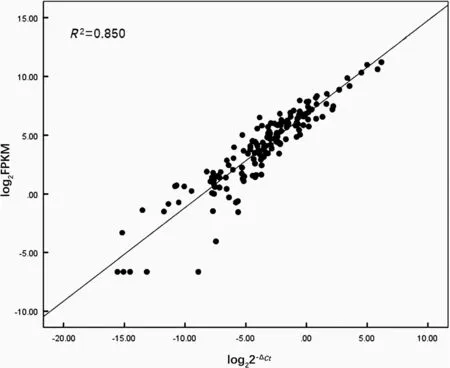
Fig.2 Comparison between gene expression level result from RNA-Seq and qRT-PCR.x-axis represents log2-transformed FPKM values obtained from RNA-Seq data,y-axis represents log2-transformed 2-ΔCTvalues calculated based on qRT-PCR results
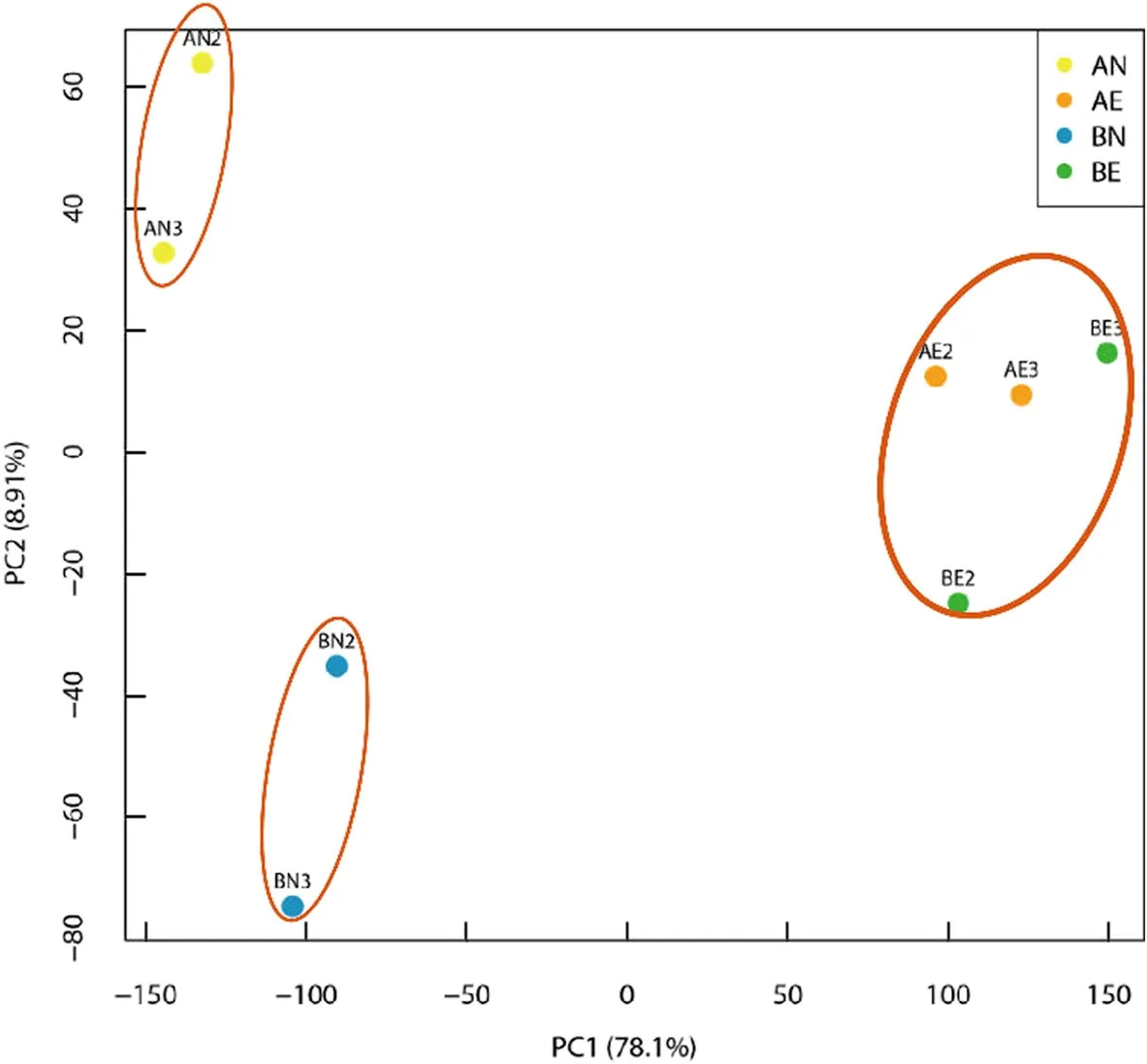
Fig.3 Principal component analysis on NEC and EC samples of CCRI 12 and CCRI 24(bad correlative duplicates were removed).NEC samples of CCRI 12 and CCRI 24 were divided clearly into two clusters,while EC samples of CCRI 12 and CCRI 24 mingled.PC1 stands for principlecomponent 1,and PC2 stands for principle component 2
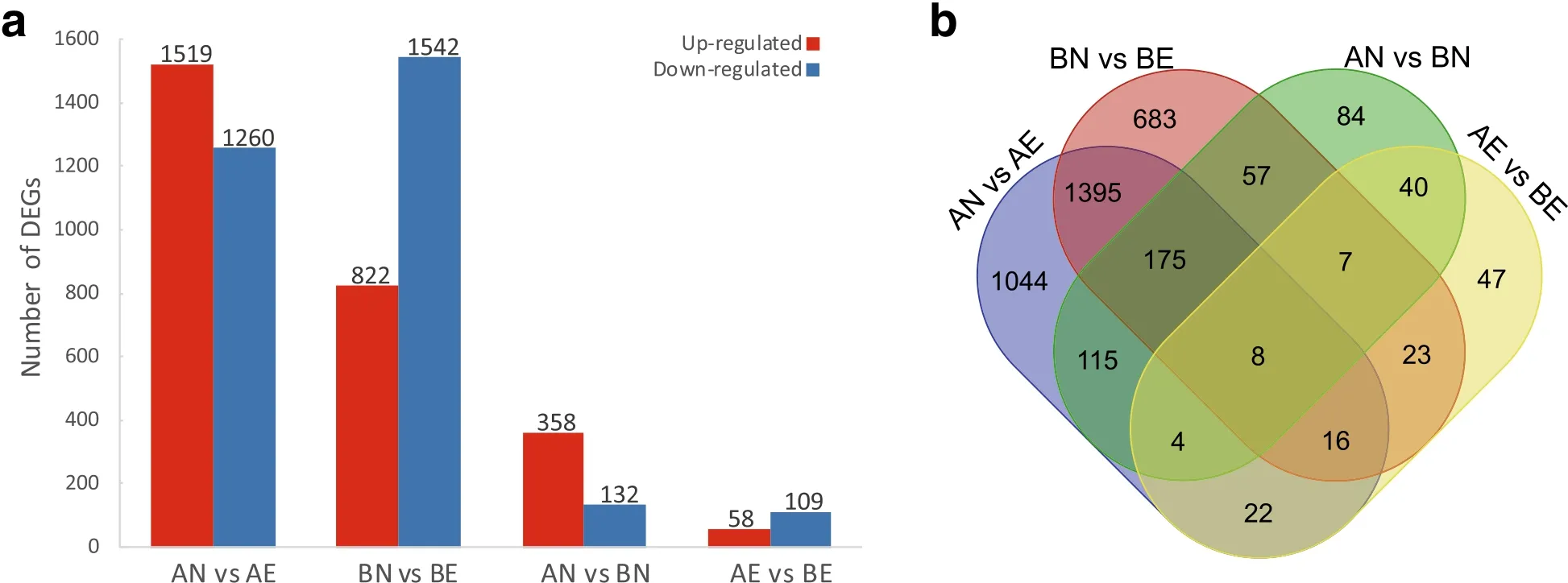
Fig.4 DEGs in CCRI 12 and CCRI 24 during transformation from NEC to EC.a The number of up-regulated and down-regulated DEGs compared between different callus states(NEC and EC)and different cultivars(CCRI 12 and CCRI 24).b Venn diagram showing overlapping regions corresponding DEGs detected in two or more comparison pairs
To compare the expression pattern of DEGs involving the NEC to EC conversion between CCRI 12 and CCRI 24,all the 3 549 DEGs detected in two cultivars were divided into three sets:Set I(1 594 DEGs)was significantly differentially expressed in both cultivars;Set II(1 185 DEGs)was significantly differentially expressed uniquely in CCRI 12;Set III(770 DEGs)was significantly differentially expressed uniquely in CCRI 24.All the three DEGs Sets were subjected to hierarchical clustering(Fig.5).Result showed that in all three Sets,most of the DEGs showed same expression pattern between CCRI 12 and CCRI 24 during the NEC to EC transition process.Especially in Set I,almost all DEGs(1 592)showed same expression pattern between two cultivars,except two paralogous genes with no annotation.Interestingly,when concerning up-regulated DEGs in Set I,CCRI 12 up-regulated with a higher ratio than CCRI 24 during the conversion process.As CCRI 12 showed relative lower expression levels of these genes in NEC than CCRI 24,higher ratio of up-regulation in CCRI 12 resulted in similar expression level in EC between two cultivars.In Set II,there were significantly more DEGs up-regulated(803)than DEGs down-regulated(382).On the contrary,the vast majority of DEGs in Set III were down-regulated(662 DEGs).
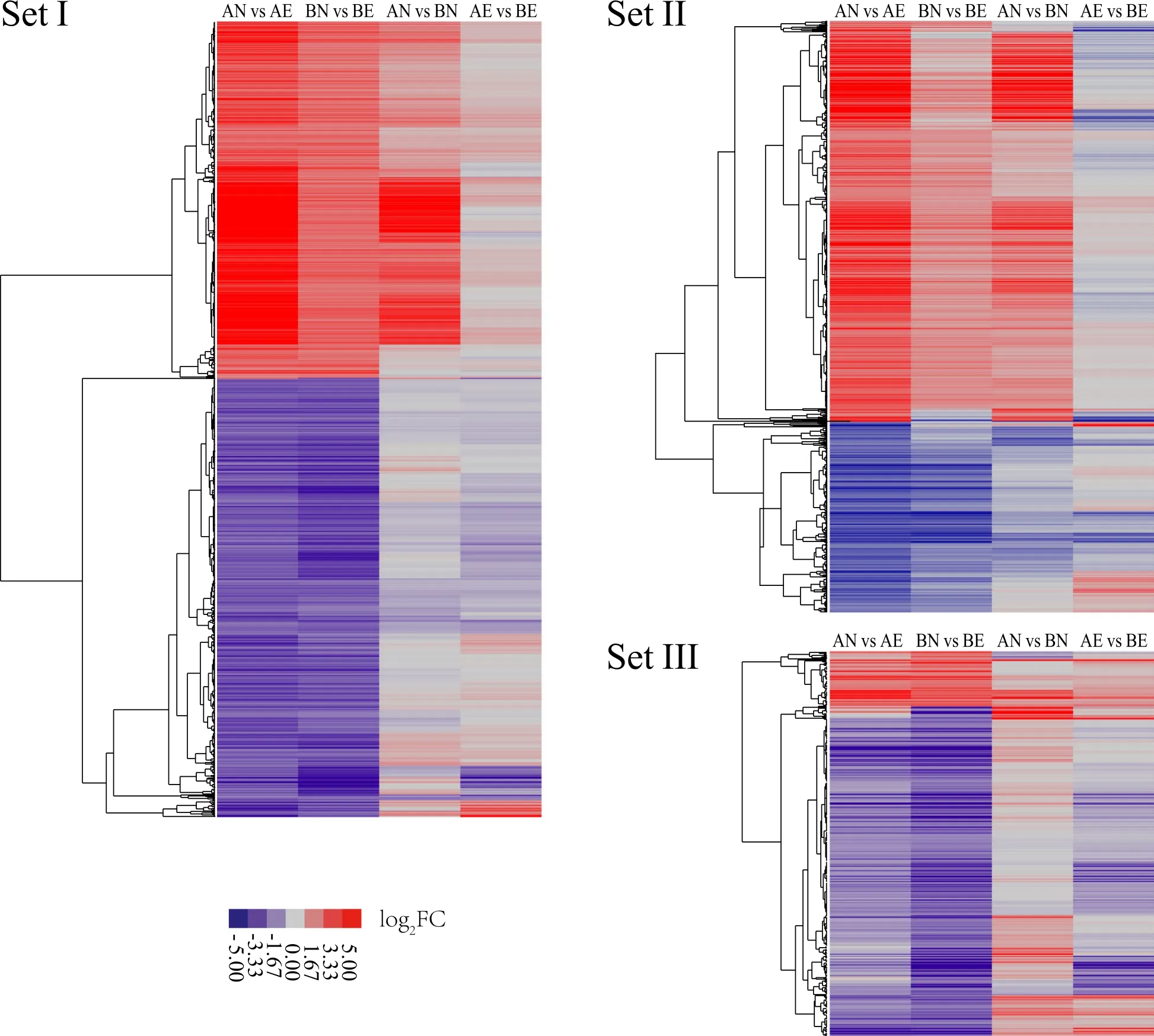
Fig.5 Schematic of expression pattern hierarchical clustering of 3 549 DEGs during transformation from NEC to EC.All DEGs were divided into three sets,DEGs in Set II showed differentially expressed uniquely in CCRI 12,DEGs in Set III showed differentially expressed uniquely in CCRI 24,DEGs in Set I showed differentially expressed in both cultivars.The log2-transformed fold change(FC,represents the ratio of FPKM in a sample type to that in another sample type)values were used to construct the heat map
Gene ontology annotations were performed on all DEGs detected during the transformation process to provide insight about their molecular characteristics and biological function.A total of 912,1 108,and 569 DEGs derived from AN vs AE comparison were annotated by biological process,molecular function,and cellular component terms,respectively.The counterpart numbers of DEGs derived from BN vs BE comparison were 764,917,and 424,respectively.Enrichment results showed that similar functional categories were enriched between CCRI 12 and CCRI 24 during the transformation from NEC to EC(Fig.6).On aspect of biological process,cellular and metabolic processes were most highly enriched.Besides,many genes involved in single-organism process,localization,and response to stimulus.Rhythmic process was uniquely enriched in CCRI 12,while immune system process was uniquely enriched in CCRI 24.The most highly enriched molecular function terms were binding and catalytic activity,which is also the molecular function term with the largest numbers of genes assigned.In the binding subset,protein binding transcription factor(TF)activity was uniquely enriched in CCRI 12,whereas nucleic acid binding transcription factor activity was uniquely enriched in CCRI 12 and CCRI 24.Under cellular component category,most of the genes were assigned to cell part,especially organelle and membrane part.Two membraneenclosed lumen related genes were uniquely enriched in CCRI 12(Fig.6).
To identify the biological pathways that are active during transformation process from NEC to EC,all DEGs detected during the transformation were annotated against Kyoto Encyclopedia of Genes and Genomes(KEGG)(Kanehisa and Goto 2000)database.As a result,1 568 DEGs in CCRI 12 were assigned to 111 pathways,1 295 DEGs in CCRI 24 were assigned to 111 pathways.Enrichment analysis result showed that the most highly enriched pathways were metabolic and secondary metabolites biosynthesis pathways(Fig.7).Flavonoid biosynthesis and phenylpropanoid biosynthesis were the most highly enriched secondary metabolites pathways.Plant hormone signal transduction pathway was enriched in CCRI 12,but not in CCRI 24,which indicated the important role of plant hormone signal transduction in the NEC to EC transition in CCRI 12.
TF encoding genes expression analysis during the NEC to EC transformation
From all the 3 549 DEGs detected in two cultivars,a total of 322 TF encoding genes were identified.257 TF encoding genes were detected in CCRI 12,about half were up-regulated and half were down-regulated.181 TF encoding genes were detected in CCRI 24,significantly more downregulated TF encoding genes were detected(Table 2).All the 322 differentially expressed TF encoding genes were assigned to 39 TF families.AP2-EREBP and MYB were the two largest TF families,which accounted for 19.3%and 15.8%of the genes,respectively.Other TF families such as bHLH,NAC,LOB,WRKY,C2H2,ABI3VP1,and C2C2 accounted for 39.4%(each accounted for 3.7%to 8.4%)of the TF encoding genes(Fig.8).When Comparing CCRI 12 with CCRI24,seven (Alfin-like,CSD,FHA,HSF,NOZZLE,RWP-RK,S1Fa-like)TF families were uniquely detected differentially expressed in CCRI 12,while three(FAR1,mTERF,Sigma70-like)families were uniquely detected differentially expressed in CCRI 24.
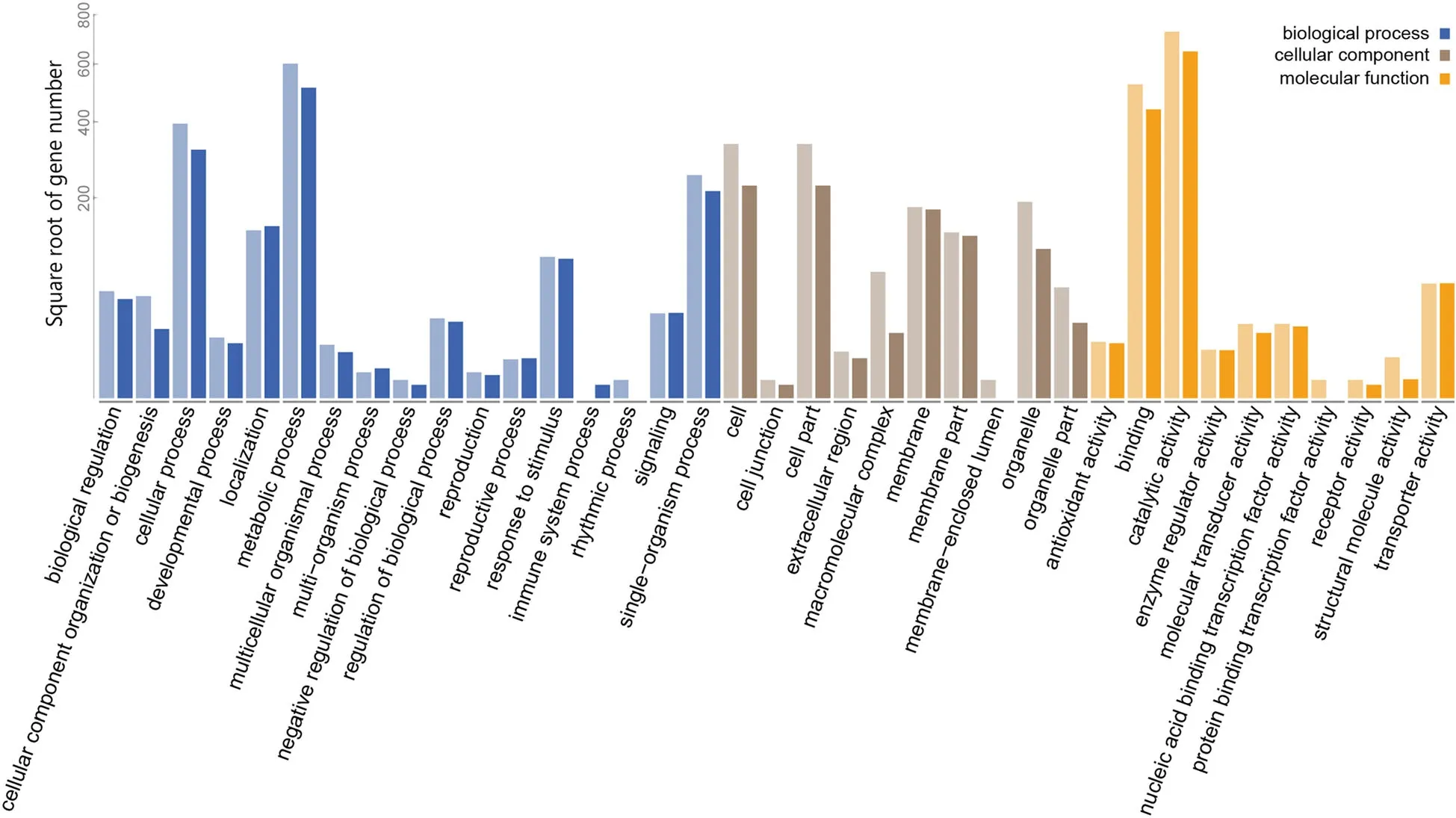
Fig.6 GO functional categories of DEGs.GO terms were classified into three main categories:biological process(blue color),cellular component(brown color)and molecular function(yellow color).Comparing CCRI 12(in deep color)with CCRI 24(in light color),two cultivars showed similar functional classification
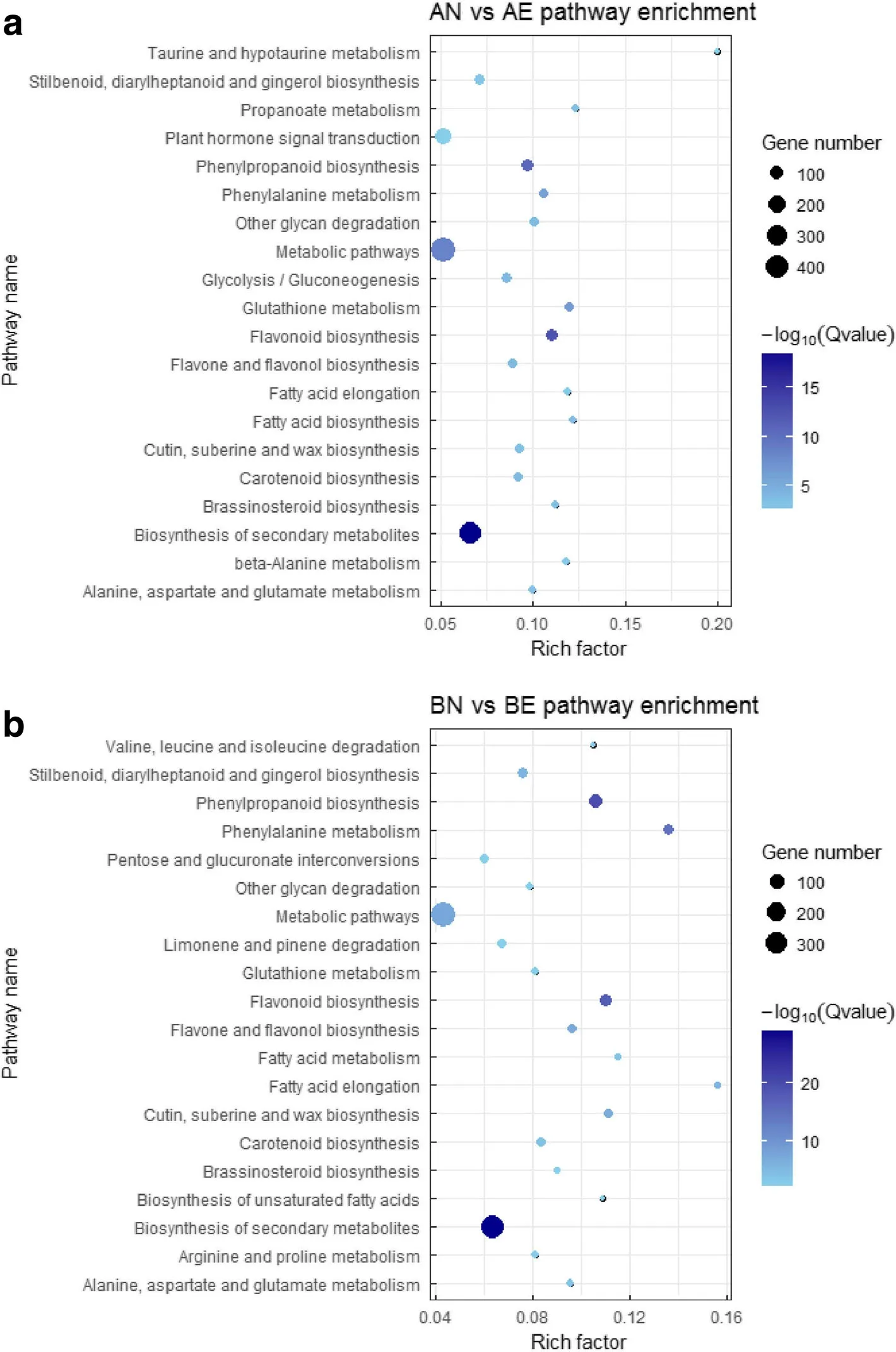
Fig.7 Top 20 enriched pathways during transformation from NEC to EC in CCRI 12(a)and CCRI 24(b)based on KEGG

Table 2 Summary of differentially expressed transcription factor(TF)encoding genes compared between two cultivars and different callus states
Discussion
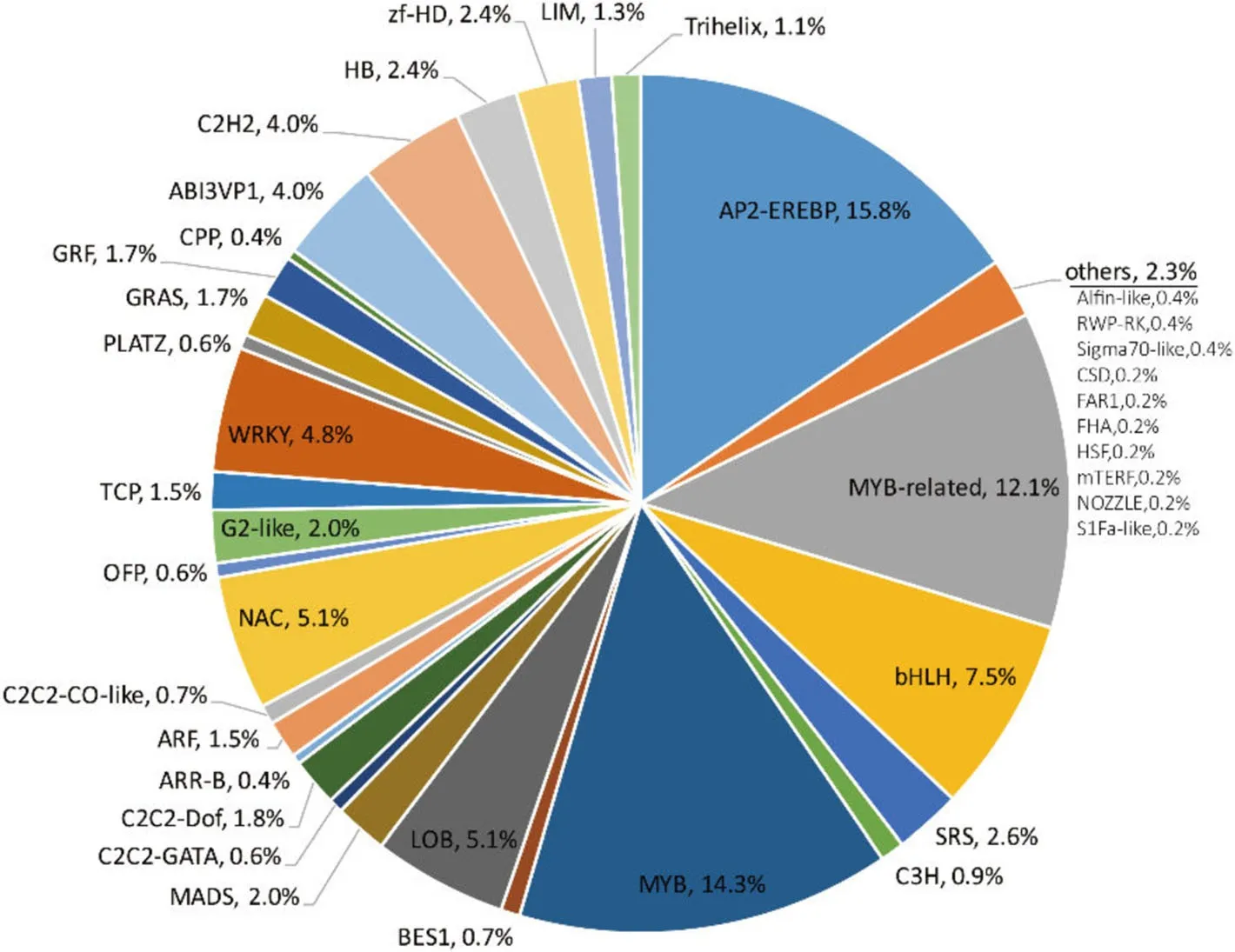
Fig.8 Gene family distribution of differentially expressed transcription factor(TF)encoding genes.Total 322 differentially expressed TF encoding genes were classified into 39 TF families.Numbers represent the percentage of the corresponding TF families.The sector“others”is constituted of seven TF families uniquely identified in DEGs of CCRI 12 and three TF families uniquely identified in DEGs of CCRI 24
Although CCRI 12 and CCRI 24 have different SE capabilities,we obtained embryogenic calli from both cultivars,suggesting that both of them have genetic potentials for SE and embryonic callus formation competence.Under the same culture conditions,different SE efficiencies of CCRI 12 and CCRI 24 are likely due to differences in gene expression(Xu et al.2013).Based on the transcriptome profile obtained by RNA sequencing,we compared gene expression patterns between NEC and EC of CCRI 12 and CCRI 24 to dissect the underlying molecular mechanism of transformation from NEC to EC,which is the key restriction in SE.In the two cultivars,most of the DEGs were implicated and enriched significantly in metabolism and secondary metabolites biosynthesis,especially flavonoid biosynthesis and phenylpropanoid biosynthesis pathways which are closely related.Flavonoids are synthesized from phenylpropanoid derivatives by condensation with malonyl-CoA(De Vries 2000;Vogt 2010;Saito et al.2013).Genes encoding key enzymes of phenylpropanoid biosynthesis,such as dihydroflavonol-4-reductase,chalcone synthase,chalcone isomerase,flavonoid 3′-hydroxylase,and flavonoid 3′,5′-hydroxylase,were down-regulated from NEC to EC.Similarly,genes encoding key enzymes in flavonoid biosynthesis,such as phenylalanine ammonialyase,cinnamate-4-hydroxylase,and 4-coumarate-CoA ligase,were also down-regulated from NEC to EC.These results are consistent with previous studies on metabolites,which reported that the content of flavonoids and phenolic compounds in EC were lower than NEC(Zhang et al.1992;Li et al.1994;Wu et al.1999).Flavonoids are a large class of plant secondary metabolites with many functions,including pigmentation,antioxidant activity,and response to biological and abiotic stresses(De Vries 2000;Saito et al.2013).Down-regulation of genes encoding key enzymse in flavonoid biosynthesis and phenylpropanoid biosynthesis may indicate a decreased secondary metabolism in EC,while major primary metabolic enhancement may favor cell differentiation(Zhang et al.1992).More of these involved genes were down-regulated in CCRI 24(120 in 137)comparing with CCRI 12(104 in 133)during the transformation from NEC to EC.For example,all the seven putative 2-oxoglutarate and Fe(II)-dependent oxygenase superfamily protein encoding genes in CCRI 24 were down-regulated,whereas three of them showed highly up-regulated(log2Ratio values were 2,4,and 9,respectively)in CCRI 12.The NAD(P)-binding Rossmann-fold superfamily protein encoding genes were up-regulated significantly in CCRI 12,while down-regulated in CCRI 24.Our results showed that flavonoid and phenylpropanoid biosynthesis processes were down-regulated more greatly in CCRI 24 than CCRI 12.More vigorous secondary metabolism in CCRI 12 may hinder the main primary metabolism required,and result in tardiness of cell differentiation and gain of embryonic callus.
Plant hormones were found to participate in the regulation of SE(Fehér et al.2003;Jiménez 2005).Exogenous plant hormones such as IAA,2,4-D,and KT are commonly added to medium to induce and regulate SE(Liu et al.2014).In the present study,from NEC to EC,the plant hormone signal transduction pathway was enriched in the recalcitrant cultivar CCRI 12,but not in CCRI 24.While comparing NEC with EC,148 plant hormone signal transduction-related genes were differentially expressed in CCRI 12,containing 77 up-regulated genes and 71 downregulated genes.One hundred nine plant hormone signal transduction-related genes were differentially expressed in CCRI 24,containing 34 up-regulated genes and 75 down-regulated genes.Concerninghormonesignal transduction-related genes,CCRI 12 showed significant more DEGs up-regulated than CCRI 24.Besides,all up-regulated DEGs showed significant higher ratio of up-regulation in CCRI 12 than CCRI 24.All of these indicate a more dramatical regulation in CCRI 12,implying that hormone signal transduction may be more crucial for the recalcitrant cultivar CCRI 12 to obtain embryonic competence than CCRI 24.This seems to contradict a previous study,which found that signal transduction of plant hormones was more likely to be the determining factor for recalcitrant cotton lines to obtain EC than biosynthesis(Xu et al.2013).
Principal component analysis showed that NEC tissues of CCRI 12 and CCRI 24 were highly heterogeneous,whereas the EC tissues of CCRI 12 and CCRI 24 were similar.It may mean that the EC tissues derived from different(even any)genotypes have similar molecular status,despite the difference of NEC.Therefore,we hypothesized that the differentially expressed transcripts detected in AE vs BE comparison were attributed to the genetic background differences between cultivars,and were unrelated to SE.While the differentially expressed transcripts detected in AN vs BN comparison were more likely to be SE-related.Of the 490 DEGs detected in the AN vs BN comparison,175 DEGs showed differential expressions during the transformation from NEC to EC in both CCRI 12 and CCRI 24(except for eight genes detected in AE vs BE comparison).All of these DEGs showed same expression pattern between CCRI 12 and CCRI 24 during the transformation,with 155 DEGs up-regulated and 20 DEGs down-regulated.All the 155 up-regulated genes had higher expression levels in BN than in AN,including five genes that were not detected in AN.The expression of these genes was positively correlated with the formation of EC,indicating that the higher expression levels of these genes in BN may account for the more effective formation of EC.Of the 20 down-regulated DEGs,13 genes had higher expression levels in AN than BN,and other 7 genes had higher expression levels in BN than AN.The expression of these genes was negatively correlated with the formation of EC.
SE marks the end of the explant gene expression pattern and the beginning of the embryonic gene expression pattern(Han et al.2009).We assumed that some transcripts may be critical or favorable for SE,deficiency of these transcripts in CCRI 12 may hinder the initiation of transformation from NEC to EC.By comparing AN with BN,we attempted to identify genes that contributed to initiation of the embryogenic transformation.Thirty transcripts were detected in BN but not in AN.Among them,one gene encoded each of the following proteins,GSTU43,disease resistance-responsive family protein,a phosphatidyltransferase,phloem protein 2-A1,nucleotide binding protein,DnaJ subfamily C member 2,a heat shock protein,WD-repeat protein,cysteine-rich protein kinase,seed storage 2S albumin superfamily protein,SNARE-like superfamily protein,and ELF4-like protein,5 genes encoded uncharacterized proteins,7 encoded hypothetical proteins(five were L484_002552),and 6 had no annotations.Most of these transcripts were up-regulated in CCRI 12,implying they may be crucial for effective embryogenesis,and accumulation of these transcripts may promote the initiation of embryogenic shift.Among these transcripts,7 transcripts,including 5 for hypothetical protein L484_002552,one for uncharacterized protein TCM_032689 and one for phosphatidyltransferase, showed opposite expression patterns,up-regulated in CCRI 12 and down-regulated in CCRI 24.We considered that these transcripts may be significantly up-regulated to activate the reprogramming for embryonic transformation and subsequently down-regulated,which implied that these transcripts may play important roles specifically in the initiation of transformation from NEC to EC.Ten up-regulated transcripts in both CCRI 12 and CCRI 24 and 6 up-regulated transcripts in CCRI 12 may be required throughout the NEC to EC transformation to maintain the transformation process.Six transcripts were only detected in CCRI 24,including four transcripts encoding cysteine-rich protein kinase,seed storage 2S albumin superfamily protein,SNARE-like superfamily protein,and ELF4-like protein respectively,and two encoding uncharacterized proteins.The transcript that encoded seed storage 2S albumin superfamily protein was uniquely detected in BN,which is interesting because this transcript showed down-regulated in BE comparing with BN,implying its specific role in initiation of transformation.This transcript may be significantly up-regulated in BN to activate the initiation of embryonic transformation,then down-regulated through the whole transformation process,as indicated by its absence in AN and AE.
In contrast,some transcripts may be repressive for SE,accumulation of these transcripts in CCRI 12 may also hinder the NEC initiate the transformation to EC.Sixteen transcripts were detected in AN but not in BN,and most of them were absent in both BN and BE.Thirteen of the transcripts existed only in CCRI 12,including one transcript that uniquely existed in AN.Among these thirteen transcripts,three were down-regulated in AN compared with AE,one encoded calcium-binding EF-hand family protein,one encoded fiber protein Fb17,and one encoded multi-vesicular body protein.It is possible that these three transcripts may account for the recalcitrance of CCRI 12,especially the fiber protein Fb17 coding one which was detected only in AN.The down-regulation of these genes may be conducive to the removal of suppression and the subsequent initiation of embryonic transformation.
In current study,during the transformation from NEC to EC,we sampled only at the initial and end points.Because many biological processes are gradual,and the transition from NEC to EC spans a long time,the two end-timepoint samplings may have missed gene expression information in the intermediate processes.For example,some transcripts may appear in a special middle period,but not at either end.Sampling at more timepoints during the process will provide more information about the gene expression dynamics that takes place from NEC to EC.More precise transcriptome profile will provide insight into more actual cellular regulation during the transformation process.
Conclusions
Comparative analysis of transcriptome profiles derived from RNA sequencing indicate that EC tissues are highly homogenous,despite between genotypes with different SE abilities.The transformation from NEC to EC involves extensive metabolism changes.In SE-recalcitrant cultivars,excessive secondary metabolism,especially flavonoid and phenylpropanoid biosynthesis,may hinder cell differentiation and delay the emergence of embryogenic attributes.Plant hormones play important roles in EC induction,and we found that the recalcitrant genotype likely had poorer ability to perceive the exogenous hormones and signaling.Take published upland cotton genomes as references,the present transcriptome analysis study provided new insight into molecular mechanism of the intractable conversion of NEC to EC in cotton,and may help for related functional genes mining.
Acknowledgements
We gratefully acknowledge Li WJ for her encourage and support.
Funding
This work was supported by National Science and Technology Major Project(2016ZX08010004),China.
Availability of data and materials
Plant materials are available from the corresponding author on reasonable request.The datasets supporting the results of this article are included within the article and its additional files.
Authors’contributions
Tian RP designed and carried out the experiment,Sun RB analyzed the data and wrote draft manuscript.Ma D and Liu CL supervised the research and gave final approval of the version to be published.All the authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Received:14 May 2018 Accepted:20 August 2018

杂志排行

Journal of Cotton Research的其它文章
- Identification and screening of nitrogenefficient cotton genotypes under low and normal nitrogen environments at the seedling stage
- Hypoxia tolerance studies for yield,fiber and physiological traits in cotton(Gossypium hirsutum L.)
- A genome-wide analysis of SWEET gene family in cotton and their expressions under different stresses
- Map-based cloning of a recessive gene v1 for virescent leaf expression in cotton(Gossypium spp.)