以感觉异常起病的Wilson病伴ATP7B基因新突变1例
2018-11-07陈君逸徐迎胜樊东升
陈君逸徐迎胜樊东升
Wilson 病(Wilson disease,WD)是一种常染色体隐性遗传疾病,因铜代谢异常致病,临床表现包括神经和精神症状、肝症状、角膜色素环等,致病基因为ATP7B基因。本文报道了一例少见的以感觉异常起病的成年WD患者,基因检查显示ATP7B基因复合杂合突变,其中母源获得p.I1284N为新突变,此前未见报道。
1 临床资料
患者女性,29岁,主因“双手麻木疼痛3个月,伴发作性右侧面部、肢体发紧感”入院。患者3个月前出现双手手指麻木、疼痛,遇冷加重,双手颜色无明显变化,症状持续约10 d缓解,伴发作性右侧面部、肢体发紧感,右侧口角上抬,持续3~4 s后可自行缓解,于发病3 d内反复发作,平均6~7次/d,每次症状相似,发作时无意识丧失、大小便失禁。此后无类似症状发作,入院时觉右足行走时疼痛明显。病程中无发热,无头晕、头痛,无视物改变,无言语不清、吞咽困难及饮水呛咳,无肢体无力。既往:5年来间断节食、口服多种减肥药(包括含有决明子、薏苡仁、泽泻、茯苓、荷叶、葛根等成分的复方中药制剂以及盐酸西布曲明等),体重于55~80 kg间波动,近3个月体重无明显变化。患者为独生女,父母否认近亲婚配,否认相关家族史。查体:身高 163 cm,体重 85 kg(BMI 31.99 kg/m2),神清,颅神经查体未及明显异常,四肢肌容积、肌张力对称正常,四肢肌力V级,双侧共济运动正常。四肢浅、深感觉正常对称。四肢腱反射正常,病理征均未引出。辅助检查:HCY 40.96 μmol/L明显升高(参考值 5~15 μmol/L),血、尿、便常规、肝肾功、电解质、心肌酶、血脂、血糖、自身免疫抗体(ANCA、ANA、抗dsDNA抗体、抗ENA谱、抗心磷脂抗体等)、感染性疾病相关(HBV、梅毒、HIV)均未见明显异常,2个月前行腰穿检查脑脊液常规、生化、神经节苷脂抗体、自身免疫脑炎相关抗体均未见明显异常;脑电图检查未见明显异常;常规神经传导检测示四肢神经传导未见异常,BAEP示双侧听觉-脑干径路传导阻滞。以“周围神经病变待查”收入院。
入院后行心电图、头MRA及CEMRV均未见明显异常;治疗后复查HCY 8.37 μmol/L正常范围,因筛查肺癌相关瘤标SCC、CYFRA21-1轻度升高,予完善胸部CT示心肺未见异常,意外发现肝脏多发略低密度灶;遂行腹部B超示脾大,肝脏实质弥漫性病变;进一步完善肝脏CT(平扫+增强)示肝内多发低强化小结节,考虑肝硬化。査头颅MRI(平扫+增强)示双侧丘脑、中脑对称性T2WI异常高信号,左基底节区多发T2WI异常高信号,增强扫描未见明显强化(图1 A1-2)结合患者病史,颅脑影像学检查示丘脑、中脑对称性病灶,累及基底节,合并肝脏病变,考虑Wilson病可能,予完善眼科检查,可见双角膜色素环(图2);血清铜蓝蛋白 124 mg/L明显下降 (参考值 250~630 mg/L),血清铜 15.4 μmol/L 正常偏低(参考值 11.8~39.3 μmol/L)。进一步行基因检查(ATP7B基因Sanger法测序):ATP7B基因exon 13:c.2924C>A,p.S975Y,错义突变,父源(图 3);exon 18:c.3851T>A,p.I1284N,错义突变,母源(图 4)。
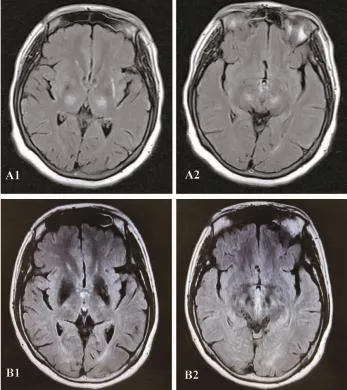
图1 头颅MRIA1、2:入院时头颅MRI T2Flair相:双侧丘脑、中脑对称性异常高信号,左基底节区多发异常高信号;B1、2:出院34个月后随访,复查头颅MRI T2Flair相:双侧丘脑、中脑对称性异常高信号及左侧基底节区异常高信号较前减小

图2 眼科裂隙灯检查可见双角膜色素环
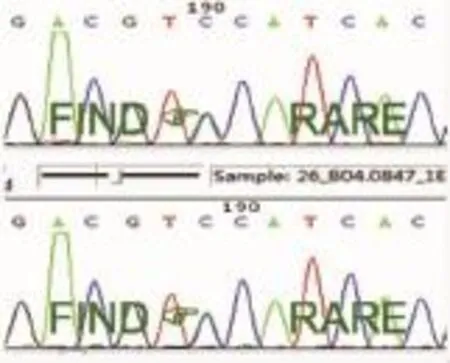
图3 ATP7B基因测序结果exon 13:c.2924C>A,p.S975Y,父源
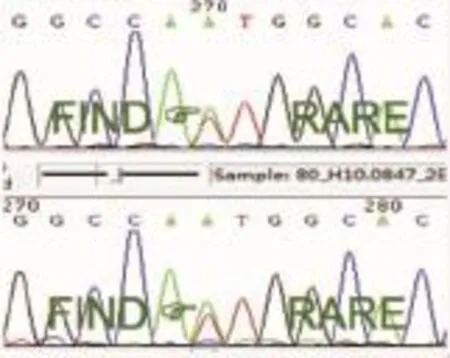
图4 ATP7B基因测序结果exon 18:c.3851T >A,p.I1284N,母源
患者青年起病,主要表现为双手麻木疼痛,伴发作性右侧面部、肢体发紧感,头颅影像学检查可见双侧丘脑、中脑对称性异常信号,左基底节区多发异常信号,血清铜蓝蛋白明显下降,血清铜正常偏低,按照公式:血清游离铜(μg/L)=63.5×血清铜(μmol/L)-3.15×铜蓝蛋白(mg/L)[1-2],估算其血清游离铜为 587.3 μg/L,明显增高>102.4 μg/L,伴角膜色素环、肝硬化,基因检查示ATP7B基因致病性复合杂合突变,根据欧洲肝脏研究协会2012年发布的WD临床指南,患者WD诊断明确。
患者出院后持续青霉胺驱铜治疗,分别于出院后3个月、6个月、14个月、26个月、34个月随访,患者未诉肢体麻木及强直感,复查HCY、血肝功、肾功、心肌酶、电解质均未见明显异常,出院后34个月复查颅脑MR可见病灶较前缩小(图1 B1、2),提示治疗效果良好。
2 讨论
Wilson病,即肝豆状核变性,是一种常染色体隐性遗传的铜代谢异常疾病,ATP7B基因为其致病基因,由于铜排泄减少导致铜在多种器官内沉积而致病。WD在世界内发病率为1/30000~1/100000,致病基因携带者约为1/90[3-4],可于任何年龄发病,大多数患者于5~35岁发病[1-2],临床表现包括神经症状(锥体外系为主)和精神症状、肝症状、角膜色素(Kayser-Fleischer ring,K-F)环、肾损害症状等。WD是少数存在有效药物治疗的神经遗传病之一,早期治疗有效,不经治疗可导致死亡[4]。
本患者首发症状为双手麻木疼痛,为WD少见表现,查体及神经传导检查未及明显异常,经治疗后症状好转,考虑周围神经感觉小纤维受累可能。以往有以肢体感觉障碍、无力等周围神经病表现起病的WD个案报道[5-6],腓肠神经活检病理提示髓鞘及轴索混合性损害,与慢性肝病及代谢性疾病所致周围神经病理改变相似[6],其发病机制尚不明确,可能与铜代谢异常相关。此外,患者发病前间断服用多种减肥药数年,包括复方中药制剂以及盐酸西布曲明等,需与减肥药物所致神经系统不良反应相鉴别。减肥药物可根据其作用部位分为作用于中枢神经系统的食欲抑制剂和作用于外周的消化吸收阻滞剂两种[7-8],前者包括芬氟拉明、右芬氟拉明、氟西汀、吗吲哚、西布曲明等,可引起包括神经系统在内的多系统不良反应。芬氟拉明和右芬氟拉明可引起恶心、腹泻、嗜睡、口干、头痛、头晕、旋转性眼震、下颌震颤等不良反应[7]。复方中药制剂中的某些成分可能对周围神经存在毒性。减肥药不良反应与患者症状相关性尚不明确,可于停药后继续观察随访以进一步鉴别诊断。
ATP7B基因为WD致病基因,编码铜转运P型ATP酶,该酶主要在肝细胞内表达,参与肝细胞铜与血清铜蓝蛋白结合及经胆道排铜[2-3]。目前已发现该基因超过600种突变,可能的致病突变超过500种,其中错义突变最为常见 (数据来自Human Gene Mutation Database及Wilson Disease Mutation Database)。大多数WD患者为复合杂合突变[2,9],世界不同地区和人种WD患者携带的ATP7B基因突变种类差异很大,欧洲人群以H1069Q突变的携带率最高,而东亚人群则以R778L突变最为常见[2-3,9]。我国WD患者 ATP7B基因有 3个突变热点:R778L、R992L和T935M,约占所有突变的60%左右[4]。
本例患者携带ATP7B基因的两个错义突变,符合复合杂合突变,其中来自父源的突变位于exon 13:c.2924C>A,p.S975Y,已被HGMD收录,2003年由GU和KODAMA等[10]报道,为我国一例汉族WD患者所携带。另一个来自母源的突变位于 exon 18:c.3851T>A,p.I1284N,为新突变(novel mutation),ExAc数据库、千人基因组数据库、HGMD及WDMD均未报告,应用Poly-Phen2、Mutation Taster等软件进行预测为致病突变(disease causing)、可能损害蛋白功能(probably damaging),在不同物种间高度保守(图 5);蛋白质功能分析显示该突变位点位于铜转运P型ATP酶ATP酶区hinge功能域,I1284N突变导致异亮氨酸被亮氨酸取代,进而可能影响蛋白结构,对ATP酶功能产生影响。综上,根据2015年ACMG针对基因变异类型的判断标准[11],I1284N 满足致病证据 PM1、PM2、PP2、PP3, 符合可能的致病突变(likely pathogenic)。

图4 变异不同物种保守性分析
WD在临床上并不少见,早期进行药物治疗患者可明显获益,但不经治疗具有较高的致死率,因此早期诊断具有很高的临床价值。但是由于WD临床表型复杂多样,部分患者在就诊时未出现K-F环等特征性临床表现,为诊断造成一定的困难。本文报道的WD患者以少见的周围神经损害表现为首发症状,成年起病,无家族史,以周围神经病变待查收入院,因筛查发现肝脏病变,继而发现颅脑影像学检查发现丘脑、中脑对称性病灶,累及基底节,进而行眼科裂隙灯检查、铜蓝蛋白及基因检查确诊,并发现可能为致病突变的ATP7B基因新突变p.I1284N,经治疗患者症状好转,颅内病灶缩小。WD可以周围神经损害为首发症状,临床医师需提高对其认识,若发现颅脑影像学对称性病灶,查体时需关注角膜有无K-F环,及时进行眼部及肝脏检查,必要时行铜蓝蛋白及基因检查,以提高WD的诊断率,及早治疗,增加患者获益。
