克唑替尼药物关键手性中间体合成进展
2018-11-06弓添添肖美娟何钰书
弓添添,肖美娟,陈 樱,何钰书,王 普
(浙江工业大学药学院,浙江 杭州 310014)
0 引言
肺癌是全世界最常见且发病率最高的恶性肿瘤。据2014年世界卫生组织报道,全球范围内每年有180万的新发病例和160万的死亡病例;在我国,随着环境污染问题日益严重,每年新发病例为67.7万,致死人数达56.5万,均为世界癌症患病率和死亡率排行榜第一[1]。肺癌分为两类:非小细胞肺癌(NSCLC,占85%左右)和小细胞肺癌(SCLC,占15%左右)[2-4]。分子靶向药物是目前治疗非小细胞肺癌的首选药物,相对于传统的化疗具有更高的选择性,且毒副作用小[5]。
克唑替尼(Crizotinib,XalkoriR),化学名称为3-[(R)-1-(2,6-二氯-3-氟苯基)乙氧基]-5-[1-(哌啶-4-基)-1H-吡唑-4-基]吡啶-2-胺,结构式见图1,相对分子质量为 450.34,分子式为C21H22Cl2FN5O,性状为白色至浅黄色粉末[6]。克唑替尼通过选择性竞争三磷酸腺苷(ATP),阻断激酶蛋白来发挥作用,使细胞停留在G1-S期,从而抑制肿瘤细胞的增殖和诱导细胞凋亡[7-8]。该药物2011年获美国FDA批准上市,成为全球第一个口服的EML4-ALK融合基因抑制剂,也推动了分子靶向药物向个体化治疗的发展。制备克唑替尼的难点和关键在于其关键手性中间体 (S)-1-(2,6-二氯-3-氟苯基)乙醇的合成。本文主要综述了克唑替尼关键手性中间体 (S)-1-(2,6-二氯-3-氟苯基)乙醇的合成方法,包括生物酶法,化学合成法,手性拆分法,酶偶联催化,挖掘新型酮还原酶催化以及微生物全细胞催化。

图1 药物克唑替尼结构式Figure 1 The structural formula of Crizotinib
1 生物酶法
Pei-Pei Kung[9]报道了以 2,6-二氯-3-氟苯乙酮(化合物1)为起始原料,通过NaBH4、甲醇还原后制得消旋体2,该消旋体2经过酯化反应得到酯类化合物3,继而利用猪肝酯酶水解酯类化合物3得到S型和R型手性中间体4。最后将得到的R型中间体4在LiOH和甲醇体系中转化为单一S构型的手性醇(图2)。该方法所得产物ee值为97%,但由于生物酶不易获得,价格昂贵,反应条件苛刻易失活,反应步骤多,周期长,收率低等限制了其进一步应用。

图2 猪肝酯酶酶法合成(S)-醇Figure 2 Synthesis of(S)-alcohol by pig liver esterase (PLE)
2 化学合成
唐虹[10]报道了化学合成方法,以2,6-二氯-3-氟苯乙酮为起始原料,在溶剂中通过有机小分子催化剂(S)-二甲苯基脯氨醇,在还原剂金属硼氢化钠和三甲基氯硅烷的作用下,经不对称还原得到(S)-1-(2,6-二氯-3-氟苯基)乙醇,收率为98%,ee值为96%,该方法克服了原有技术中采用生物酶催化步骤多,周期长和化学拆分法收率低等缺点,具有成本低、操作简便、收率高、光学纯度好等优点,适用于大规模生产。
Qian Jian-Qiang等[11]报道了利用 Ir[(R)-DTB-spiro-PAP-3-Me]铱吡啶氨基膦化氢为催化剂,以2,6-二氯-3-氟苯乙酮为起始原料,在叔丁醇钾,无水乙醇和氢气体系下不对称催化合成(S)-1-(2,6-二氯-3-氟苯基)乙醇(见图 3),该方法的转化率高,ee值为99.5%,但铱催化剂(CAS:1418483-59-6)无市售,因此限制了该合成路线的应用。
3 手性拆分
孙学英[12]等采用Boc-L-脯氨酸(N-叔丁氧基羰基-L-脯氨酸)与催化剂对甲苯磺酸和缩合剂1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐相组合的催化拆分方法制得S型产物。该路线以2,6-二氯-3-氟苯乙酮(化合物6)为起始原料,经水解制得消旋体1-(2,6-二氯-3-氟苯基)乙醇(化合物7),将消旋体(化合物7)拆分为S-型醇和R-型醇(化合物8),随后将拆分副产物混合物(化合物8,化合物9,化合物 10)水解得到S-型醇和R-型醇(化合物11),而后将R-型醇(化合物11)与三苯基磷和偶氮二甲酸二乙酯相组合选择性的进行Mitsunobu反应,产物(化合物12)再经过氢氧化钠水解,结晶分离提纯制得(S)-1-(2,6-二氯-3-氟苯基)乙醇(见图4),缩短了反应时间,减少了污染,易于工业化实施,但总收率较低。
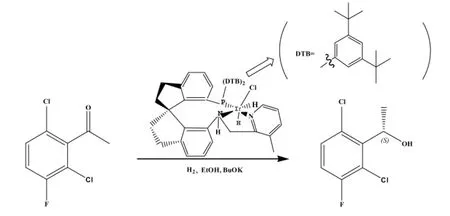
图3 金属催化制备(S)-醇Figure 3 Synthesis of(S)-alcohol by metal as catalyst
4 酶偶联法催化
郭红颜等[13]利用基因工程的方法,以洛德酵母(Lodderomyces sp.)基因组为模板,经PCR扩增得到甲酸脱氢酶(FDH)基因,导入载体pET28a,构建重组质粒pFDH-pET28a,并构建乙醇脱氢酶(ADH) 表达质粒 pADH-pET21a,与 pFDH-pET28a 共 同 转 入 E.coli BL21 (DE3)(S)-1-(2,6-二氯-3-氟苯基)乙醇(见图 5),产物纯度为100%,收率50%以上。
刘丽勤[14]构建重组大肠杆菌E.coli BL21-ADH和E.coli BL21-GDH,实现葡萄糖脱氢酶和乙醇脱氢酶的共表达,进行酶偶联转化(图6)。结果表明,在反应温度为 30℃,pH=7下,(S)-1-(2,6-二氯-3-氟苯基)乙醇的产量最高,在投料量为6%时,转化率为93.75%。

图4 手性拆分制备(S)-醇Figure 4 Synthesis of(S)-alcohol by chiral resolution

图5 乙醇脱氢酶偶联甲酸脱氢酶制备(S)-醇Figure 5 Synthesis of(S)-alcohol by coupling alcohol dehydrogenase and formate dehydrogenase

图6 共表达乙醇脱氢酶和葡萄糖脱氢酶制备(S)-醇Figure 6 Synthesis of(S)-alcohol by coexpression of alcohol dehydrogenase and glucose dehydrogenase
5 新型酮还原酶的挖掘
魏东芝[15]以2,6二氯-3-氟苯乙酮为底物进行酶的筛选,从酶库中筛选到可完全转化底物且产物为S构型手性醇,ee值大于99.5%的开菲尔乳酸菌酮还原酶(KRED82,Lactobacillus)(图 7)。当底物浓度为3 M (621 g/L)时,反应24 h后底物转化率可达99.5%。
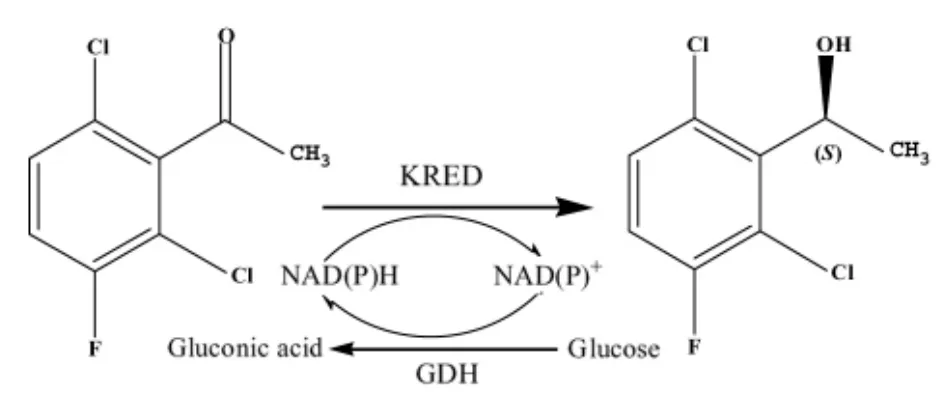
图7 利用酮还原酶合成(S)-醇Figure 7 Synthesis of(S)-alcohol by ketoreductase
6 微生物全细胞催化
笔者所在实验室[16]筛选得到一株白地霉(Geotrichum candidum)ZJPH1704, 可用于高选择性催化底物2,6-二氯-3-氟苯乙酮合成 (S)-1-(2,6-二氯-3-氟苯基)乙醇。底物浓度 1 g/L,200 g/L菌体量(湿重),于 40℃下反应 24 h,产率达93%,ee值大于99.9%。利用全细胞催化具有反应条件温和,催化剂性能稳定且不易失活,选择性好等优点。
7 结论与展望
近年来,随着人们的物质生活水平快速提升,癌症的发病率也逐年提高,肺癌作为发病率和致死率第一的癌症,对人类的健康存在巨大威胁。克唑替尼是目前非小细胞肺癌的首选治疗药,有着较好的市场前景,因此其研究开发上市和制备技术的发展受到越来越多的关注。目前对于克唑替尼的制备技术虽然已较为成熟但仍为化学方法合成,反应过程中存在副产物多,步骤较为繁琐,环境不友好等问题,所以在克唑替尼的制备技术开发方面仍值得研究。利用生物催化法制备克唑替尼具有高效、光学纯度高、环境友好,以及其在化合物合成上能避免一些化学法所必须的苛刻条件等优势,使得生物催化法受到广泛地关注,如何将生物法更好地运用于克唑替尼制备的工艺路线中是未来研究的方向。
