适于陆地棉品种身份鉴定的SNP核心位点筛选与评价
2018-11-06朱国忠李乐晨牛二利郭旺珍
朱国忠 张 芳 付 洁 李乐晨 牛二利 郭旺珍
适于陆地棉品种身份鉴定的SNP核心位点筛选与评价
朱国忠 张 芳 付 洁 李乐晨 牛二利 郭旺珍*
南京农业大学 / 作物遗传与种质创新国家重点实验室 / 杂交棉创制教育部工程研究中心, 江苏南京 210095
利用全基因组SNP信息, 筛选陆地棉品种特异的核心SNP位点组合, 可为陆地棉品种身份鉴定提供准确高效的检测手段。本研究利用棉花CottonSNP80K芯片对326份不同来源的陆地棉种质进行SNP分型。以南京农业大学陆地棉TM-1基因组(AD1) genome NBI v1.1版本为参考序列, 对SNP位点进行注释。结果表明, 93.85% (72 990/77 774)的位点检出率超过99%, 61 595 (79.20%)个SNP位点具有多态性, 其中76.32% (47 009)的位点最小等位基因频率(MAF)大于0.1。基于位点检出率大于0.99、位点具多态性、MAF大于0.2、杂合率小于0.05、每条染色体的SNP密度为400 kb/SNP左右等要求, 最终获得4857个覆盖全基因组的高质量核心SNP位点组合。这些核心SNP位点组合平均检出率接近100%; 平均MAF值为0.34; 平均杂合率为0.02; 99%以上的陆地棉材料均能够被准确鉴定。统计分析表明利用核心SNP位点组合与CottonSNP80K的鉴定结果呈极显著相关。本研究提供了包含4857个SNP位点, 适于陆地棉品种指纹图谱绘制的核心SNP位点组合, 可实现陆地棉品种身份鉴定和品种确权。
DNA芯片; 指纹图谱; SNP; 核心位点; 陆地棉
棉花(spp.)是世界性重要的天然纤维作物, 也是重要的油料作物之一, 在农业生产和国民经济中占有重要地位。陆地棉和海岛棉是两大异源四倍体栽培棉种。陆地棉产量高, 适应性广, 纤维品质中等, 占全球棉花种植面积的95%以上[1]。中国是世界最大的棉花生产、消费和纺织大国, 在棉花种植和新品种培育方面有悠久的历史和丰富的经验[2]。然而, 中国非棉花起源国, 生产上种植的陆地棉栽培品种遗传背景狭窄。随着育种进程的加快, 少数骨干亲本的集中应用, 以及转基因等新技术在棉花育种中的应用, 棉花品种间的遗传多样性越来越小[3-4]。再加上品种套种等销售过程的不规范现象, 仅仅依靠形态性状越来越难辨别棉花品种, 给种子生产、经营、管理、维权等诸多环节带来挑战, 急需从DNA水平研制准确表明每一品种身份的“指纹”。
基于各种类型的分子标记, 很多研究者已经开展标记技术在棉花品种鉴定中的应用[5-6]。其中SSR (Simple Sequence Repeats)分子标记以重复性好、操作简单、呈共显性等优点, 应用最为广泛。利用SSR标记技术, 武耀廷等绘制了4个高优势杂交种的指纹图谱[7], 马轩等[8]建立了18个彩色棉品系的DNA指纹图谱, 秦利等[9]开展了新疆部分主栽品种的指纹图谱构建和杂交种纯度鉴定研究。赵亮等通过全基因组范围SSR标记筛选, 获得一套多态性高, 易于鉴别, 且涉及四倍体陆地棉26条连锁群的26对SSR引物, 并用于棉花品种DNA条形码数据库构建[10]。上述研究对棉花品种身份及纯度鉴定起到了一定的作用。但相比于全基因组覆盖的SNP (Single Nucleotide Polymorphism)标记, SSR标记仍存在基因组中分布不均匀、基因分型多态性不高, 以及基因组覆盖度不够等问题, 尚不能满足不同基因型高通量分子鉴定及选择的需求。SNP标记是基因组中分布最广泛, 数量最丰富的DNA分子标记。利用覆盖全基因组的SNP芯片, 可以通过一次杂交实现数以万计、十万计、乃至百万计的SNP位点分型, 具有成本低、通量高、获得的信息量大等优势, 受到广泛的关注和应用[11-12]。匡猛等[13]基于KASP技术开发了一套适用于我国棉花杂交纯度检测的核心SNP技术, 为棉花指纹图谱构建提供支撑。孙正文等[14]利用CottonSNP63K芯片筛选了393个核心SNP位点, 构建了719份陆地棉资源材料的特征DNA指纹图谱。
结合芯片技术, 筛选覆盖全基因组的核心SNP位点, 绘制陆地棉栽培品种SNP指纹图谱, 开展自动化、高通量、准确的棉花种子质量监控研究, 可为棉花种业安全提供保障。前期本研究室公布了较高质量的异源四倍体棉种陆地棉遗传标准系TM-1基因组信息[15], 结合100余份不同来源陆地棉材料的重测序信息, 研发了覆盖全基因组、适于陆地棉种内基因型鉴定的高多态SNP芯片(CottonSNP80K), 该芯片位点源于陆地棉种内差异, 可寻址, 多态性高, 能有效用于陆地棉种内基因型鉴定和目标基因发掘[16]。本研究利用CottonSNP80K芯片对300余份不同来源陆地棉品种/材料的分型结果, 通过分析不同位点遗传多样性鉴定信息, 筛选高多态、可重复、易分辩、强专一的SNP位点集合, 用于构建陆地棉品种指纹图谱和品种确权分析, 服务种子产业化和品种知识产权保护需求。
1 材料与方法
1.1 试验材料
共326份, 包括遗传背景广泛, 不同来源的陆地棉品种/材料312份(编号C21-C332), 和不同的近等基因系14份(编号E7-E20)。材料信息及编号与Cai等[16]一致。所有资源材料引自中国农业科学院棉花研究所国家棉花种质中期库, 由南京农业大学棉花研究所自交保存。
1.2 DNA提取与质量控制
针对每一供试材料, 选取健康种子发芽, 生长到二叶一心时, 摘取棉花嫩叶, 采用CTAB法提取基因组DNA[17]。用1%琼脂糖电泳检测所提样品的DNA质量, 要求DNA条带单一、无弥散、无RNA残留。利用One Drop-OD 1000进一步检测DNA质量和浓度, 要求光吸收值260/280介于1.7~2.1、样品浓度 > 50 ng μL–1、单个样品总量 > 1 μg。
1.3 SNP检测与数据分析
利用我们自主研发的棉花CottonSNP80K芯片[16], 进行供试材料的基因组SNP分型分析。参照标准实验流程(基于光纤微珠芯片的Infinium技术, the Illumina protocols)检测SNP, 用iScan芯片扫描仪对杂交结果扫描。委托北京康普森生物技术有限公司完成全基因组扫描。将通过芯片扫描获得的原始数据导入GenomeStudio软件(V2011.1, Illumina, Inc.)进行SNP分型。由于GenomeStudio只能以二倍体模式分型, 针对异源四倍体陆地棉, 我们在默认分型的结果上进行了人工调整, 使分型结果更加准确[16]。SNPs的位置对应南京农业大学陆地棉TM-1基因组(AD1) genome NBI v1.1版本的参考基因组序列信息[15]。软件分型后自动输出SNP位点检出率。根据SNP位点的分型结果, 使用Microsoft Excel软件计算与统计分析最小等位基因频率(MAF)、杂合率和多态信息含量(PIC)。
1.4 SNP位点分型结果验证
使用在线工具WebSNAPER (pga.mgh.harvard. edu/cgi-bin/snap3/websnaper3.cgi)对拟选择验证的SNP位点进行特异性引物设计。首先随机选择芯片检测有多态的棉花材料TM-1、I4005、DPL15、泗棉3号等对所开发的SNP引物进行多态性筛选, 进一步选择不同来源的24份陆地棉材料进行SNP位点的验证分析。统计比较聚丙烯酰胺凝胶电泳(PAGE)与芯片的分型结果, 评价芯片的分型准确率。SNP-PCR扩增的反应程序为, 95℃ 5 min预变性; 94℃ 30 s变性, 65℃ 30 s退火, 72℃30 s延伸, 28循环数; 72℃终延伸10 min。扩增产物经非变性PAGE电泳(恒压226 V, 1.0~1.5 h), 通过银染DNA显带, 记录多态性数据。
2 结果与分析
2.1 SNP位点特性评估
前期我们研发了覆盖全基因组、适于陆地棉种内基因型鉴定的高多态SNP芯片(CottonSNP80K), 并利用该芯片获得300余份不同来源陆地棉品种/材料的基因组分型结果[16]。为了进一步发掘核心SNP位点, 用于陆地棉栽培品种指纹图谱构建, 统计分析了CottonSNP80K芯片中所有位点的检测结果(图1)。CottonSNP80K中包含77 774个有效的SNP位点, 63.61% (49 477)的SNP位点间的距离在2~10 kb, 15.05% (11 706)的位点间距大于30 kb, 标记的染色体密度为24.9 kb/SNP。利用该芯片对312份陆地棉材料(编号: C21~C332)进行SNP分型, 99.40% (77 304)的位点检出率大于95%, 93.85% (72 990)的位点检出率超过99%, 表明该芯片对陆地棉SNP位点有很高的分型效率。73.02% (56 672)的位点杂合率低于5%, 但有14.89% (11 554)的位点杂合率高于50%。高杂合率的位点主要源于异源四倍体棉花A、D亚基因组间杂合。61 595 (79.20%)个SNP位点显示多态性, 其中76.32% (47 009)的位点MAF大于0.1。在此基础上进一步筛选和简化, 获得适于陆地棉品种分型的核心SNP位点组合。

图1 CottonSNP80K芯片SNP位点特征统计
横坐标代表被统计的SNP特征参数, 依次为位点检出率、最小等位基因频率、杂合率和相邻SNP之间的距离。纵坐标代表SNP的分布数目。
The abscissa represents statistical SNP characteristic parameters, involved in loci call frequency, minor allele frequency (MAF), heterozygosity and distance between adjacent SNPs. The ordinate represents the number of SNPs.
2.2 用于品种指纹图谱构建的核心SNPs挖掘
基于CottonSNP80K中77 774个SNP位点的多态性和相邻SNP间距信息, 进一步开展分层筛选, 确定核心SNP位点。(1)去除检出率小于99%的SNP位点后, 余72 990个位点; (2)去除供试材料中表现SNP单态的位点, 余57 244个位点; (3)筛选MAF大于0.2的位点, 获得28 851个位点; (4)筛选杂合率小于0.05的位点, 获得19 934个位点; (5)基于前期研究提供的陆地棉栽培品种连锁不平衡(LD)距离[16], 标记密度选择平均每条染色体400 kb/SNP, 分型相对简单, 具有二倍体作物SNP特性。结合人工过滤, 删除由多拷贝产生的复杂分型位点, 确保核心位点的特异性, 最终获得4857个多态性高、重复性好、二维表型易分辩、特异性强的高质量SNP位点。
表1显示, 中选的4857个SNP位点中, 2908个位于A亚组, 分布密度为399.0 kb/SNP; 1949个位于D亚组, 分布密度为397.3 kb/SNP。由于陆地棉群体LD衰减距离约为700 kb[16], 本研究筛选的核心SNP位点其分布密度可以覆盖全基因组。
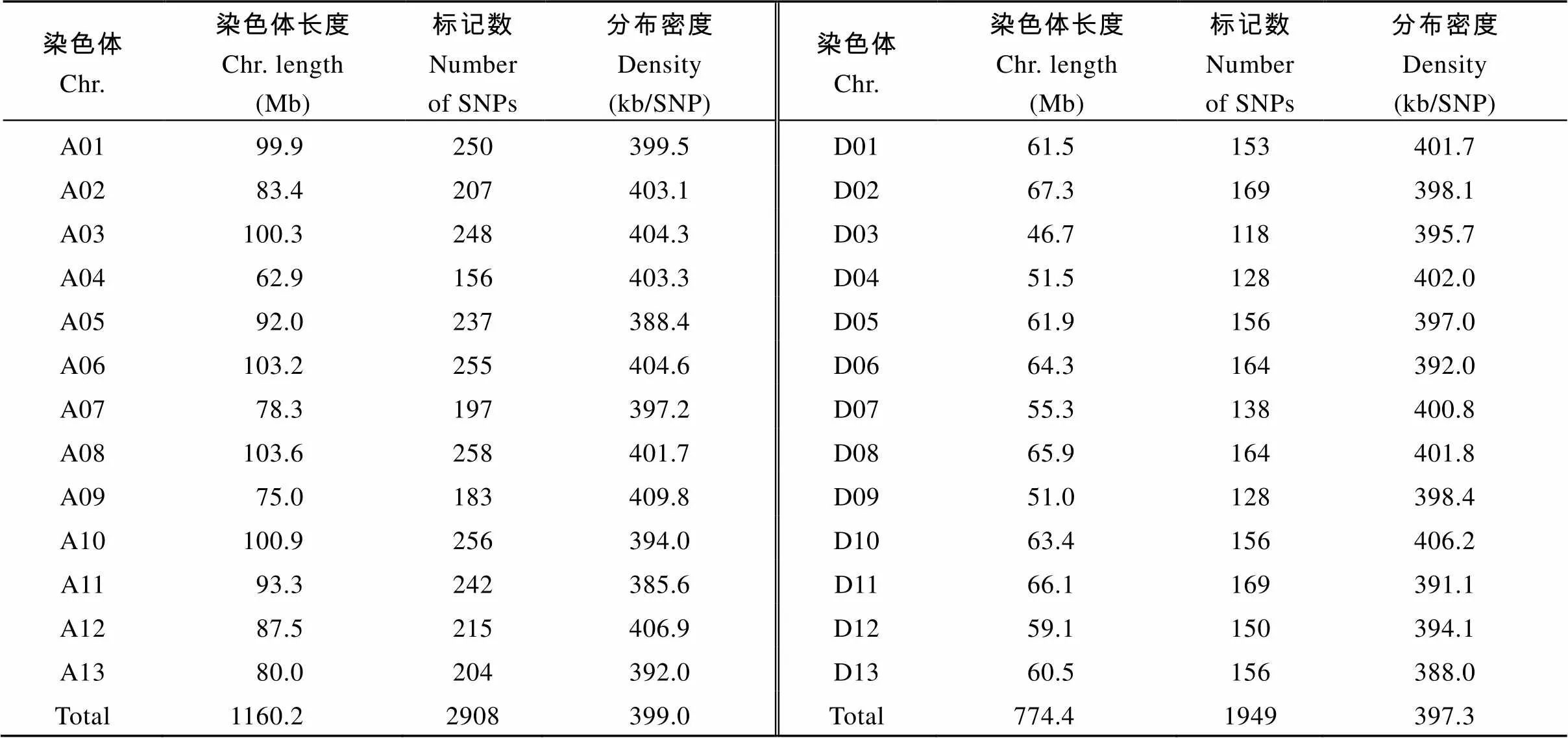
表1 用于指纹图谱绘制的核心SNP位点染色体分布
4857个候选SNP位点的各参数与CottonSNP80K上的原始位点检测结果比较, 本研究用于品种指纹图谱分析的核心位点检出率接近100%; 平均MAF值为0.34, 平均PIC为0.44, 多态性显著提高; 同时, 候选位点的平均杂合率降低到0.02, 更加有利于陆地棉品种身份及真实性检测(表2)。
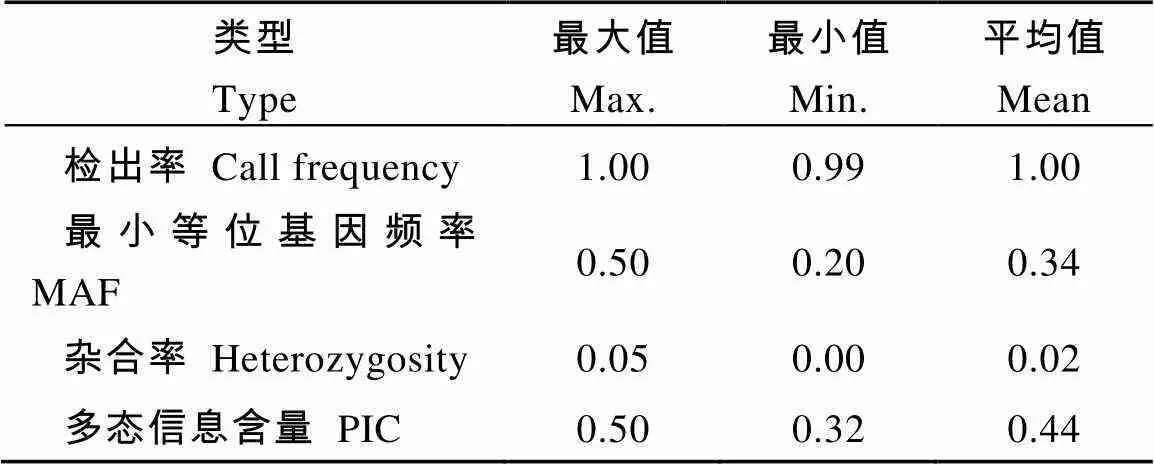
表2 用于品种指纹图谱分析的核心SNP位点特征统计
2.3 核心SNP组合鉴定陆地棉栽培品种效率评估
使用核心SNP位点组合对312份陆地棉品种/材料进行身份鉴定, 99%以上的品种均能实现准确有效的鉴别, 其遗传距离最大为0.6047, 最小为0.0001。为了评估候选SNP对不同陆地棉品种的鉴定效率, 分别用CottonSNP80K总77 774位点和用于指纹图谱绘制的4857个位点对312份陆地棉品种材料进行遗传距离相关性分析(图2), 结果显示, 利用核心SNP位点组合与CottonSNP80K的鉴定结果呈极显著线性相关(< 0.01)。表明本研究所筛选的核心SNP位点集合具有代表性, 可有效用于陆地棉品种身份鉴定。
为了进一步评估核心SNP位点组合的分辨力, 选择14份近等基因系材料(编号: E7~E20)进行遗传多样性分析。基于核心位点SNP检测结果, 通过两两比较, 获得近等基因系材料之间的差异位点数(多态位点)并计算其占总位点的比例(多态率)。与CottonSNP80K芯片上77 774个SNP位点的检测结果相比[16], 遗传背景相近的近等基因系间其多态率均显著提高(表3)。其中新乡小吉和新乡小吉无绒无絮突变体间的多态率由16.1%提升到25.0%; 徐州142和徐州142无绒无絮突变体间由19.0%提升到30.6%; 7235和7235纤维突变体间由25.0%提升到52.4%。另外陆地棉遗传标准系TM-1与其5个纤维发育突变体间的多态性范围从22.7%~30.6%提高到44.7%~64.2%。上述分析表明, 新筛选的4587个核心SNP位点多态性高, 非常适用于陆地棉品种指纹图谱的绘制和身份鉴定。
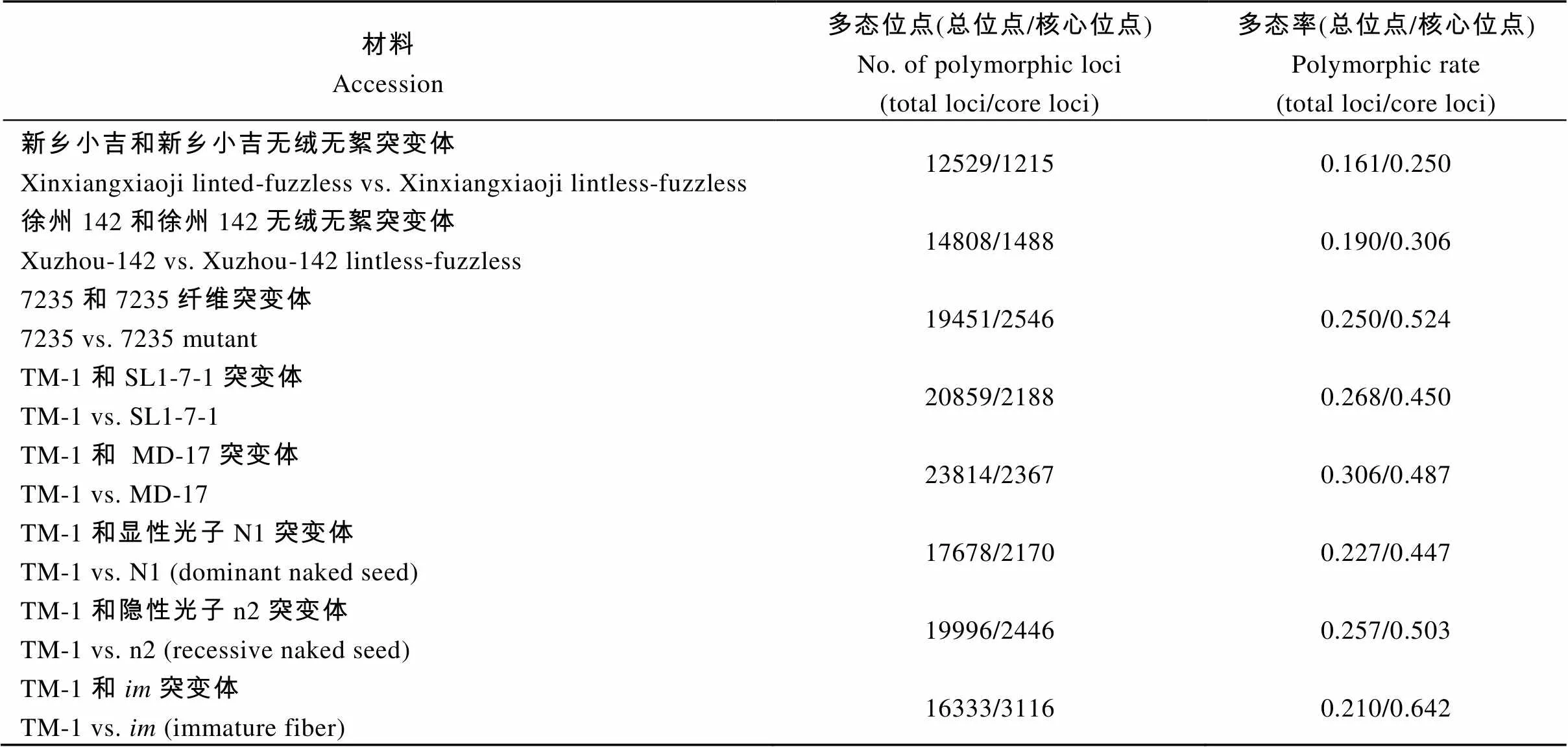
表3 基于核心SNP位点的陆地棉近等基因系多态性检测
总位点: 数据来自CottonSNP80K中77 774个位点的分析结果; 核心位点: 数据来自筛选后4857个核心位点的分析结果。
The analysis from total loci was performed using a total of 77 774 loci in CottonSNP80K, and the analysis from core loci was performed using 4857 core loci screened.
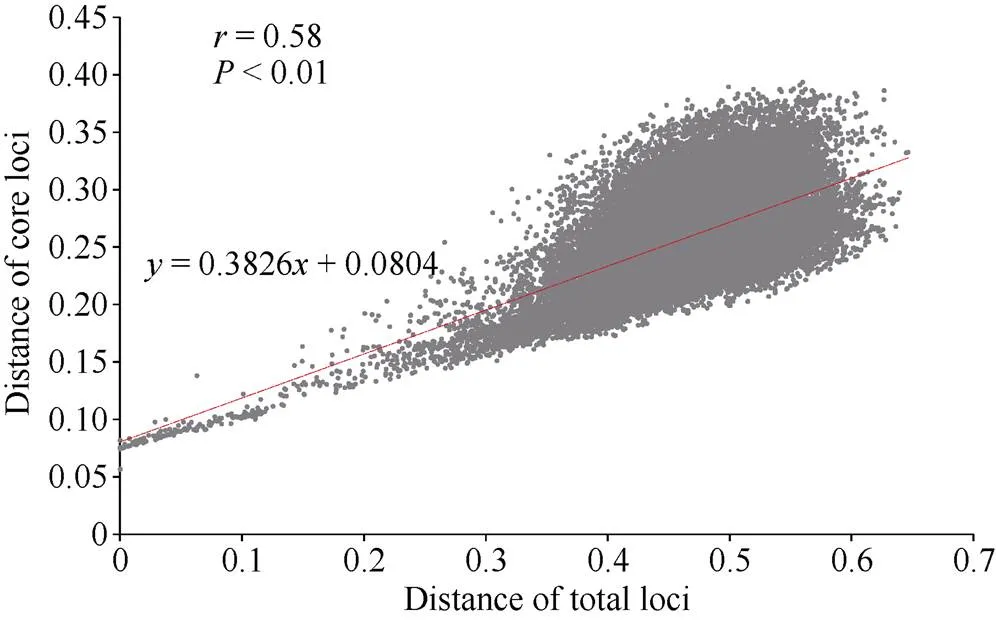
图2 312份陆地棉品种遗传距离相关性分析
横坐标代表利用芯片总位点计算的材料间遗传距离, 纵坐标代表利用核心位点计算的材料间遗传距离。
The abscissa represents the genetic distance between the materials calculated by the total loci in CottonSNP80K. The ordinate represents the genetic distance between the materials calculated by the core loci.
为了验证核心SNP位点的准确性, 从核心SNP位点中随机挑选了14个SNP位点开发特异性SNP引物, 选择24份陆地棉材料进行SNP位点和芯片分型结果的验证分析。芯片分型结果与SNP-PCR一致性高达98.8% (表4和图3)。 进一步证明这些核心SNP位点的可利用性和基于芯片SNP分型的准确性。
3 讨论
随着分子技术的不断发展与完善, 及国家不断加强对种子的管理, 作物品种指纹图谱的构建及身份鉴定技术的提升势在必行。中国是世界最大的棉花生产、消费和纺织大国, 但并非棉花起源国, 棉花的生产和育种都是从引进品种发展起来的。陆地棉原产于中美洲, 1865年最先被引入上海种植。直到20世纪20年代, 我国才开始陆地棉育种工作[18]。加上早期的引进品种有限, 使得中国的陆地棉栽培品种遗传背景十分狭窄, 利用传统的形态鉴别方法区分不同棉花品种十分困难, 迫切需要一套高精度的指纹图谱。棉花指纹图谱的绘制研究较早, 目前已经研发出许多基于SSR标记的指纹图谱, 并在某些特定棉花品种纯度及真实性鉴定方面取得较好结果[7-10]。但是由于标记数目限制, 往往不能区分遗传背景相似的品种。同时, 诸多SSR指纹图谱适用范围不同, 难以形成统一的鉴定标准。此外, 在大批量的品种检测过程中, 低通量标记技术检测也消耗大量的人力和时间, 直接影响检测效率。因此, 自动化和信息化的品种鉴定方法是棉花种业发展的必然趋势, 而高通量测序和基因芯片技术的成熟, 大大推动了棉花品种身份精准鉴定的进程。

表4 芯片分型与SNP-PCR一致性分析
最近几年, 棉花基因组序列解析及应用取得显著进展。2012年, 美国Paterson实验室牵头, 联合国际多家研究单位, 开展棉花基因组多倍化及纤维发育研究, 同时释放了二倍体D基因组雷蒙德氏棉种全基因组序列信息[19]。中国在不同倍性、不同栽培棉种的基因组信息解析上取得了突出进展。中国农业科学院棉花研究所先后于2012年、2014年公布了二倍体雷蒙德氏棉(D基因组)、亚洲棉(A基因组)全基因组序列信息[20-21]; 南京农业大学和中国农业科学院棉花研究所于2015年分别公布了异源四倍体棉种陆地棉遗传标准系TM-1基因组信息[15,22]; 溢达集团和华中农业大学于2015年分别公布了海岛棉新海21及3-79的基因组信息[23-24]。以TM-1基因组为参考序列, 南京农业大学、华中农业大学和河北农业大学分别通过对不同来源的棉花材料的全基因组重测序分析, 结合多年多点的主要农艺性状表型鉴定, 发掘出一批与棉花品种改良相关的产量、纤维品质、黄萎病抗性等关联位点, 揭示了从早期美棉品种到全球最大纤维作物改良过程中的遗传基础和驯化特征, 为棉花“精准育种”提供了优异的基因资源和理论指导[3-4,25]。
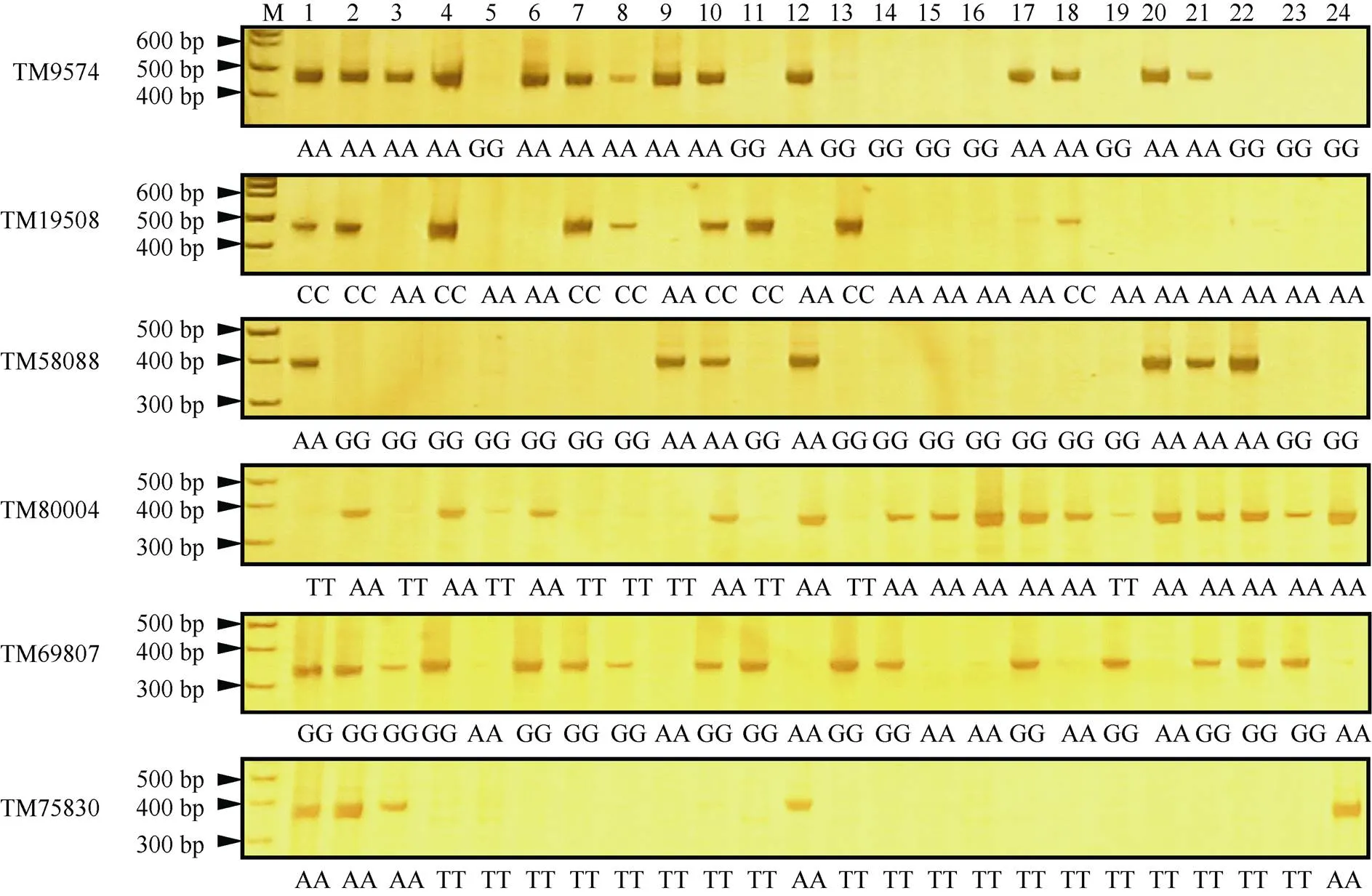
图3 基于SNP-PCR技术验证SNP位点芯片分型结果
M: DNA marker; 1: TM-1; 2: 7235; 3: J02-508(7-50); 4: 岱字棉16; 5: 鄂棉21; 6: 鄂棉23; 7: 国欣棉9; 8: 邯郸885; 9: 黑山棉1号; 10: 冀122; 11: 军棉1号; 12: 山农棉8号; 13: 山西W1; 14: 山西W8; 15: 斯字棉2B; 16: 泗棉3号; 17: 皖棉17; 18: 新陆早32; 19: 新陆早7号; 20: 新陆中26; 21: 新陆中35; 22: 豫棉15; 23: 中棉所12; 24: 中棉所41。
M: DNA marker; 1: TM-1; 2: 7235; 3: J02-508 (7-50); 4: DPL16; 5: Emian 21; 6: Emian 23; 7: Guoxinmian 9; 8: Handan 885; 9: Heishanmian 1; 10: Ji 122; 11: Jummian 1; 12: Shannongmian 8; 13: Shanxi W1; 14: Shanxi W8; 15: Stoneville 2B; 16: Simian 3; 17: Wanmian 17; 18: Xinluzao 32; 19: Xinluzao 7; 20: Xinluzhong 26; 21: Xinluzhong 35; 22: Yumian 15; 23: Zhongmiansuo 12; 24: Zhongmiansuo 41.
借鉴棉花参考基因组, 通过分析不同来源陆地棉材料的重测序信息, 我们研发了覆盖全基因组、适宜于陆地棉种内基因型鉴定的高多态SNP芯片(CottonSNP80K芯片), 并用于陆地棉栽培品种遗传多样性分析[16]。在此基础上, 本研究通过严格的条件控制最终筛选出适用于陆地棉品种检测的核心位点组合。通过陆地棉不同品种基因型区分能力评估以及位点分型真实性评估, 证明了筛选获得的核心位点组合具有高效的陆地棉品种鉴定能力。与孙正文等[14]开发的SNP指纹图谱具有97%的陆地棉品种鉴定能力相比, 我们的研究显示出多方面的优势。基于已完成的陆地棉参考基因组TM-1的序列[15], 本研究选择的SNP标记具有高检出率、单拷贝、全基因组覆盖和位点可寻址等优点。针对供试品种, 准确鉴定效率超过99%, 对遗传背景相近的品种也具有较好的鉴别能力。另外本研究广泛的基因组覆盖度使得指纹图谱的鉴定和组合具有更高的扩展性, 对已育成品种及未来育成的新品种身份鉴定均有很好的应用价值。
由于遗传背景狭窄, 陆地棉品种鉴定难度要远远高于其他多数作物。仅仅靠表型或一至两套DNA指纹图谱, 难以准确鉴定出不同来源、遗传背景不清的陆地棉栽培种。同时, 鉴定品种数目和范围的不同, 所选择的方法也有所差别。例如, 鉴定少数几个棉花品种或杂交种, 可以根据本SNP集合针对材料特点选择特异SNP位点, 通过KASP或定点测序技术鉴定品种, 或结合多态性SSR标记组合鉴定; 开展大范围的棉花品种身份鉴定, 可以通过低密度的SNP芯片绘制指纹图谱; 而对遗传背景相似的品种, 则需要绘制高密度SNP指纹图谱或开发品种特异性SNP标记, 达到深度鉴定的目的。另外, 随着育种技术的发展, 未来的新品种鉴定也需要拓宽现有技术手段。因此, 多技术多方法的组合式鉴定是陆地棉身份鉴定的核心, 同时在快速发展的育种技术背景下, 也需要进一步开发新技术新方法。
4 结论
基于CottonSNP80K芯片的陆地棉品种/材料分型结果筛选出4857个覆盖全基因组的高质量核心SNP位点组合, 该组合适于陆地棉品种指纹图谱绘制, 可实现陆地棉品种身份鉴定和品种确权。
[1] Chen Z J, Scheffler B E, Dennis E, Triplett B A, Zhang T, Guo W, Chen X, Stelly D M, Rabinowicz P D, Town C D, Arioli T, Brubaker C, Cantrell R G, Lacape J M, Ulloa M, Chee P, Gingle A R, Haigler C H, Percy R, Saha S, Wilkins T, Wright R J, Van Deynze A, Zhu Y, Yu S, Abdurakhmonov I, Katageri I, Kumar P A, Mehboob Ur R, Zafar Y, Yu J Z, Kohel R J, Wendel J F, Paterson A H. Toward sequencing cotton () genomes., 2007, 145: 1303–1310
[2] 喻树迅, 范术丽. 我国棉花遗传育种进展与展望. 棉花学报, 2003, 15: 120–124 Yu S X, Fan S L. The evolutions and prospect of cotton genetics and breeding in China., 2003, 15: 120–124 (in Chinese with English abstract)
[3] Fang L, Wang Q, Hu Y, Jia Y, Chen J, Liu B, Zhang Z, Guan X, Chen S, Zhou B, Mei G, Sun J, Pan Z, He S, Xiao S, Shi W, Gong W, Liu J, Ma J, Cai C, Zhu X, Guo W, Du X, Zhang T. Genomic analyses in cotton identify signatures of selection and loci associated with fiber quality and yield traits., 2017, 49: 1089–1098
[4] Wang M, Tu L, Lin M, Lin Z, Wang P, Yang Q, Ye Z, Shen C, Li J, Zhang L, Zhou X, Nie X, Li Z, Guo K, Ma Y, Huang C, Jin S, Zhu L, Yang X, Min L, Yuan D, Zhang Q, Lindsey K, Zhang X. Asymmetric subgenome selection and-regulatory divergence during cotton domestication., 2017, 49: 579–587
[5] 郭旺珍, 张天真, 潘家驹, 何金龙. 我国棉花主栽品种的RAPD指纹图谱研究. 农业生物技术学报, 1996, 4: 29–34 Guo W Z, Zhang T Z, Pan J J, He J L. Analysis of RAPD fingerprinting on main cotton cultivars in China., 1996, 4: 29–34 (in Chinese with English abstract)
[6] Abdalla A M, Reddy O U K, El-Zik K M, Pepper A E. Genetic diversity and relationships of diploid and tetraploid cottons revealed using AFLP., 2001, 102: 222–229
[7] 武耀廷, 张天真, 郭旺珍, 殷剑美. 陆地棉品种SSR标记的多态性及用于杂交种纯度检测的研究. 棉花学报, 2001, 13: 131–133Wu Y T, Zhang T Z, Guo W Z, Yin J M. Detecting polymorphism among upland cotton (L.) cultivars and their roles in seed purity of hybrids with SSR markers., 2001, 13: 131–133 (in Chinese with English abstract)
[8] 马轩, 杜雄明, 孙君灵. 18个彩色棉品系的SSR指纹分析. 植物遗传资源学报, 2003, 4: 305–310 Ma X, Du X M, Sun J L. SSR fingerprinting analysis on 18 colored cotton lines., 2003, 4: 305–310 (in Chinese with English abstract)
[9] 秦利, 李冰, 范玲, 李磊, 胡保民, 王沛政. 新疆陆地棉SSR标记指纹图谱构建和杂种纯度鉴定研究. 新疆农业科学, 2005, 42: 399–401 Qin L, Li B, Fan L, Li L, Hu B M, Wang P Z. Analysis on esteblishment of finger printing of SSR mark for upland cotton and purity of hybrid seed in Xinjiang., 2005, 42: 399–401 (in Chinese with English abstract)
[10] 赵亮, 蔡彩平, 梅鸿献, 郭旺珍. 用于区别不同棉花品种基因组特征的微卫星位点筛选. 作物学报, 2012, 38: 1810–1817 Zhao L, Cai C P, Mei H X, Guo W Z. Screening of microsatellite loci for identifying genome barcoding of cotton cultivars., 2012, 38: 1810–1817 (in Chinese with English abstract)
[11] McNally K L, Bruskiewich R, Mackill D, Buell C R, Leach J E, Leung H. Sequencing multiple and diverse rice varieties. Connecting whole-genome variation with phenotypes., 2006, 141: 26–31
[12] Ganal M W, Altmann T, Roder M S. SNP identification in crop plants., 2009, 12: 211–217
[13] 匡猛, 王延琴, 周大云, 马磊, 方丹, 徐双娇, 杨伟华, 魏守军, 马峙英. 基于单拷贝SNP标记的棉花杂交种纯度高通量检测技术. 棉花学报, 2016, 28: 227–233 Kuang M, Wang Y Q, Zhou D Y, Ma L, Fang D, Xu S J, Yang W H, Wei S J, Ma Z Y. High-throughput genotyping assay technology for cotton hybrid purity based on single-copy SNP markers., 2016, 28: 227–233 (in Chinese with English abstract)
[14] 孙正文, 匡猛, 马峙英, 王省芬. 利用CottonSNP63K芯片构建棉花品种的指纹图谱. 中国农业科学, 2017, 50: 4692–4704 Sun Z W, Kuang M, Ma Z Y, Wang X F. Construction of cotton variety fingerprints using CottonSNP63K array., 2017, 50: 4692–4704 (in Chinese with English abstract)
[15] Zhang T, Hu Y, Jiang W, Fang L, Guan X, Chen J, Zhang J, Saski C A, Scheffler B E, Stelly D M, Hulse-Kemp A M, Wan Q, Liu B, Liu C, Wang S, Pan M, Wang Y, Wang D, Ye W, Chang L, Zhang W, Song Q, Kirkbride R C, Chen X, Dennis E, Llewellyn D J, Peterson D G, Thaxton P, Jones D C, Wang Q, Xu X, Zhang H, Wu H, Zhou L, Mei G, Chen S, Tian Y, Xiang D, Li X, Ding J, Zuo Q, Tao L, Liu Y, Li J, Lin Y, Hui Y, Cao Z, Cai C, Zhu X, Jiang Z, Zhou B, Guo W, Li R, Chen Z J. Sequencing of allotetraploid cotton (L. acc. TM-1) provides a resource for fiber improvement., 2015, 33: 531–537
[16] Cai C, Zhu G, Zhang T, Guo W. High-density 80 K SNP array is a powerful tool for genotypingaccessions and genome analysis., 2017, 18: 654
[17] Paterson A H, Brubaker C L, Wendel J F. A rapid method for extraction of cotton (spp.) genomic DNA suitable for RFLP or PCR analysis., 1993, 11: 122–127
[18] 黄滋康. 中国棉花品种及其系谱(修订本). 北京: 中国农业出版社, 2007 Huang Z K. Cotton Varieties and Their Genealogy in China (revised and enlarged edition). Beijing: China Agriculture Press, 2007 (in Chinese)
[19] Paterson A H, Wendel J F, Gundlach H, Guo H, Jenkins J, Jin D, Llewellyn D, Showmaker K C, Shu S, Udall J, Yoo M J, Byers R, Chen W, Doron-Faigenboim A, Duke M V, Gong L, Grimwood J, Grover C, Grupp K, Hu G, Lee T H, Li J, Lin L, Liu T, Marler B S, Page J T, Roberts A W, Romanel E, Sanders W S, Szadkowski E, Tan X, Tang H, Xu C, Wang J, Wang Z, Zhang D, Zhang L, Ashrafi H, Bedon F, Bowers J E, Brubaker C L, Chee P W, Das S, Gingle A R, Haigler C H, Harker D, Hoffmann L V, Hovav R, Jones D C, Lemke C, Mansoor S, ur Rahman M, Rainville L N, Rambani A, Reddy U K, Rong J K, Saranga Y, Scheffler B E, Scheffler J A, Stelly D M, Triplett B A, Van Deynze A, Vaslin M F, Waghmare V N, Walford S A, Wright R J, Zaki E A, Zhang T, Dennis E S, Mayer K F, Peterson D G, Rokhsar D S, Wang X, Schmutz J. Repeated polyploidization ofgenomes and the evolution of spinnable cotton fibres., 2012, 492: 423–427
[20] Wang K, Wang Z, Li F, Ye W, Wang J, Song G, Yue Z, Cong L, Shang H, Zhu S, Zou C, Li Q, Yuan Y, Lu C, Wei H, Gou C, Zheng Z, Yin Y, Zhang X, Liu K, Wang B, Song C, Shi N, Kohel R J, Percy R G, Yu J Z, Zhu Y X, Wang J, Yu S. The draft genome of a diploid cotton., 2012, 44: 1098–1103
[21] Li F, Fan G, Wang K, Sun F, Yuan Y, Song G, Li Q, Ma Z, Lu C, Zou C, Chen W, Liang X, Shang H, Liu W, Shi C, Xiao G, Gou C, Ye W, Xu X, Zhang X, Wei H, Li Z, Zhang G, Wang J, Liu K, Kohel R J, Percy R G, Yu J Z, Zhu Y X, Wang J, Yu S. Genome sequence of the cultivated cotton., 2014, 46: 567–572
[22] Li F G, Fan G Y, Lu C R, Xiao G H, Zou C S, Kohel R J, Ma Z Y, Shang H H, Ma X F, Wu J Y, Liang X M, Huang G, Percy R G, Liu K, Yang W H, Chen W B, Du X M, Shi C C, Yuan Y L, Ye W W, Liu X, Zhang X Y, Liu W Q, Wei H L, Wei S J, Huang G D, Zhang X L, Zhu S J, Zhang H, Sun F M, Wang X F, Liang J, Wang J H, He Q, Huang L H, Wang J, Cui J J, Song G L, Wang K B, Xu X, Yu J Z, Zhu Y X, Yu S X. Genome sequence of cultivated Upland cotton (TM-1) provides insights into genome evolution., 2015, 33: 524–530
[23] Liu X, Zhao B, Zheng H J, Hu Y, Lu G, Yang C Q, Chen J D, Chen J J, Chen D Y, Zhang L, Zhou Y, Wang L J, Guo W Z, Bai Y L, Ruan J X, Shangguan X X, Mao Y B, Shan C M, Jiang J P, Zhu Y Q, Jin L, Kang H, Chen S T, He X L, Wang R, Wang Y Z, Chen J, Wang L J, Yu S T, Wang B Y, Wei J, Song S C, Lu X Y, Gao Z C, Gu W Y, Deng X, Ma D, Wang S, Liang W H, Fang L, Cai C P, Zhu X F, Zhou B L, Chen Z J, Xu S H, Zhang Y G, Wang S Y, Zhang T Z, Zhao G P, Chen X Y.genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites., 2015, 5: 14139
[24] Yuan D J, Tang Z H, Wang M J, Gao W H, Tu L L, Jin X, Chen L L, He Y H, Zhang L, Zhu L F, Li Y, Liang Q Q, Lin Z X, Yang X Y, Liu N A, Jin S X, Lei Y, Ding Y H, Li G L, Ruan X A, Ruan Y J, Zhang X L. The genome sequence of Sea-Island cotton () provides insights into the allopolyploidization and development of superior spinnable fibres., 2015, 5: 17662
[25] Ma Z Y, He S P, Wang X F, Sun J L, Zhang Y, Zhang G Y, Wu L Q, Li Z K, Liu Z H, Sun G F, Yan Y Y, Jia Y H, Yang J, Pan Z E, Gu Q S, Li X Y, Sun Z W, Dai P H, Liu Z W, Gong W F, Wu J H, Wang M, Liu H W, Feng K Y, Ke H F, Wang J D, Lan H Y, Wang G N, Peng J, Wang N, Wang L R, Pang B Y, Peng Z, Li R Q, Tian S L, Du X M. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield., 2018, 50: 803–813
Genome-wide Screening and Evaluation of SNP Core Loci for Identification of Upland Cotton Varieties
ZHU Guo-Zhong, ZHANG Fang, FU Jie, LI Le-Chen, NIU Er-Li, and GUO Wang-Zhen*
State Key Laboratory of Crop Genetics & Germplasm Enhancement / Hybrid Cotton R&D Engineering Research Center (the Ministry of Education) / Nanjing Agricultural University, Nanjing 210095, Jiangsu, China
Utilizing the genome-wide SNP information to screen the core SNP loci may provide an accurate and efficient method for the identification of upland cotton varieties. Using the CottonSNP80K array, SNP genotyping was performed within 326 upland cotton accessions. Then, the SNP loci were annotated with TM-1 genomic sequence of(AD1) genome NBI v1.1 Upland cotton of Nanjing Agricultural University as reference sequence. Statistical analysis of all loci in CottonSNP80K showed that the call rate of 93.85% loci (72 990 in 77 774) was more than 99%, and 61 595 (79.20%) SNPs were polymorphic loci among the tested upland cotton accessions. Among them, minor allele frequency (MAF) of 76.32% (47 009) loci was greater than 0.1. Based on call frequency for each locus > 0.99; loci with polymorphism; MAF > 0.2; heterozygosity rate < 0.05; SNP density with ~400 kb/SNP in each chromosome, we obtained 4857 high-quality core SNP loci. The characteristic statistics of the core SNP loci combination showed that the average call rate was nearly 100%; the average MAF was 0.34; and the average heterozygosity was 0.02. Using these core SNPs, more than 99% of the materials could be identified accurately and effectively. In addition, the identification results of core SNP loci showed extremely significant linear correlation with that of CottonSNP80K. Taken together, a core combination containing 4857 SNP loci for fingerprint identification of upland cotton varieties is constructed, which can accurately identify the purity and reality of modern upland cotton varieties.
DNA array; fingerprint; SNP; core loci; Upland cotton
2018-02-25;
2018-08-20;
2018-09-04.
10.3724/SP.J.1006.2018.01631
通信作者(Corresponding author): 郭旺珍, E-mail: moelab@njau.edu.cn
E-mail: zhugz@njau.edu.cn
本研究由国家重点研发计划项目(2017YFD0102000)和江苏现代作物生产协同创新中心(No.10)项目资助。
This study was supported by the National Key R&D Program for Crop Breeding (2017YFD0102000) and Jiangsu Collaborative Innovation Center for Modern Crop Production Project (No.10).
URL: http://kns.cnki.net/kcms/detail/11.1809.S.20180903.1521.008.htmll
