CRISPR/Cas9方法失活白念珠菌CaMIT1基因及其突变株表型分析
2018-11-01徐慧慧蒋伶活
徐慧慧 蒋伶活,2
(1.江南大学生物工程学院 粮食发酵工艺与技术国家工程实验室,无锡 214122;2.山东理工大学农业工程与食品科学学院,淄博 255000)
白念珠菌(Candidaalbicans)是一种人体共生菌,对免疫功能缺陷的患者能够引起系统感染,严重时会导致患者死亡[1-2]。白念珠菌感染宿主细胞时,会遭遇到宿主细胞的防御压力(例如,发热、氧化应激,以及亚硝化作用),环境压力(例如,肠道中的低氧和唾液与上皮层中的抗真菌多肽)。这些压力直接作用于白念珠菌的细胞表面。真菌表面的细胞壁是一种动态的结构,对维持细胞形态,保护细胞免受环境压力的破坏十分重要。和其他酵母细胞一样,白念珠菌的细胞壁由糖骨架组成,主要成分是β-葡聚糖,几丁质和甘露聚糖。β-1,3 和 β-1,6葡聚糖组成一个网络结构,被磷酸肽键甘露聚糖(phosphopeptidomannan, PPM)、甘露聚糖蛋白(mannoproteins)和磷脂甘露聚糖(phospholipomannan, PLM)组成的复合糖外层包围[3-4]。磷酸肽键甘露聚糖、甘露聚糖蛋白和磷脂甘露聚糖统称为甘露聚糖缀复合物,这些寡聚甘露糖缀复合物在白念珠菌与宿主的关系中发挥重要作用,与白念珠菌毒力紧密相关[5-8]。寡聚甘露糖通过β-1,2 键连接,形成一种独特的空间构象,能够被宿主的免疫系统识别。甘露聚糖通过由β-1,2连接的甘露糖残基与磷脂连接形成PLM。白念珠菌释放的PLM中的β-寡聚甘露糖可以与宿主细胞作用,通过宿主细胞上的Toll-like receptor-2诱导TNF-α合成[9-11]。尽管目前对β-甘露聚糖在白念珠菌病理生理学中的功能有诸多认识,但是对β-甘露聚糖基化的调控尚不清楚白念珠菌的β-甘露糖基转移酶(mannosyltransferase)参与β-甘露聚糖链的合成[12]。CaMIT1编码GDP-甘露糖﹕肌醇-磷酸-酰胺(inositol-phospho-ceramide, IPC)甘露糖转移酶[13]。CaMIT1突变株缺少PLM,IPCs不能够被甘露糖基化形成Mannose IPC (MIPC ),从而影响白念珠菌毒力。此外,该研究还发现CaMIT1突变株对钙离子敏感。为了进一步研究Mit1p在钙离子稳态调控中的作用,我们利用一种新的基因编辑手段CRISPR去构建CaMIT1基因的突变株。
CRISPR(Clusters of Regularly Interspaced Short Palindromic Repeats)体系,已被运用到真菌、植物和动物的研究领域中[14-15]。Fink教授实验室基于这项技术的原理,构建了在白念珠菌中可以操作的CRISPR体系,证明这种方法可以有效地编辑白念珠菌基因组[16]。白念珠菌CRISPR体系,由白念珠菌兼容性的Cas9核酸酶和一个合成的导向RNA(synthetic guide RNA)组成,sgRNA能够与基因组上特异性的20个碱基的序列进行杂交,sgRNA识别的序列紧跟着一个由NGG组成的PAM(Protospacer-Adjacent Motif)位点,Cas9蛋白能够识别PAM位点并在此位点进行剪切,因此sgRNA具有指导Cas9蛋白到基因组特定PAM位点的功能。CRISPR体系可以通过一步转化法获得两个等位基因同时失活的纯合子突变株,为基因功能和毒力研究提供了一种简单而快速的手段。以CaMIT1基因为例,我们成功通过这种方法获得了两个等位基因同时失活的CaMIT1纯合子突变株,并通过倍比稀释对Mit1p的功能进行了初步分析。
1 材料
1.1 菌株、质粒和引物
本研究所用到菌株和质粒见表1,引物见表2。
1.2 培养基
YPD培养基:20g/L蛋白胨、20g/L葡萄糖、10g/L酵母提取物,固体培养基灭菌前加入20g/L琼脂粉;YPD培养基含药物:YPD培养基湿热灭菌后,加入相应浓度的药物;LB+氨苄霉素培养基:10g/L蛋白胨,10g/L氯化钠,5g/L酵母提取物,调整pH到7.0,灭菌后加入100 μg/mL 氨苄霉素,固体培养基灭菌前加入20g/L琼脂粉。
1.3 主要试剂

表1 菌株和质粒

表2 实验所用引物序列
酵母转化用Salmon Sperm DNA (ssDNA)、LiAc、PEG 3350, 以及细胞裂解酶lyticase等试剂购自Sigma公司;所用药物诺尔斯菌素(nourseothricin, NAT)、Taq DNA聚合酶、限制性内切酶、T4连接酶等试剂购NEB公司。荧光增白剂(CFW)、酮康唑(KCZ)、氟康唑(Flu)和特比萘芬(Teb)等购自Sigma公司;氯化钙,氯化理和SDS购自国药集团。
1.4 主要仪器及设备
PCR反应仪(德国艾本德公司)、全温摇瓶柜(太仓强乐实验设备厂)、台式冷冻离心机(日本日立公司)、立式压力蒸汽灭菌锅(上海博迅实业有限公司医疗设备厂)、核酸电泳仪(北京六一仪器厂)、凝胶成像系统(美国Bio. Rad)、酶标仪(美国Bio-Rad公司)。
2 方法
2.1 引物设计
需要设计三对引物。(1)sgRNA引物设计。sgRNA是由20bp碱基组成的序列。Vyasetal. 2015的补充数据中包含有不同的sgRNA序列,选择文件(Targ.NoTs.subs12nt.HitsGenesOnly.
Hits1Gene2 Alle.3letterName),也可以利用软件Benchling进行查找。sgRNA有以下两个标准:a) sgRNA应在目的基因ORF内,且靠近起始密码子ATG,尽早终止有功能蛋白质的合成;b) On-Target和Off-Target分数越高越好,分数高表明sgRNA结合的特异性强。sgRNA需要连接到质粒pV1093上,质粒pV1093用BsmBI进行酶切后暴露出黏性末端,需在引物两端添加黏性末端,正向引物为5’-atttgX20g-3’(表2,MIT1-sgF),反向引物为5’-aaaacX20c-3’ (表2,MIT1-sgR),PAM位点应该在sgRNA的3’端(如图1C)。(2)修复模板DNA(Repair DNA)引物设计。修复模板DNA含有突变位点和同源臂,根据需要进行修饰。在构建突变株时,通过碱基修饰导入终止密码子,有3点需要修饰的地方:a) 至少一个终止密码子(UAA, UAG 或UGA),且该终止密码子必须在基因ORF中;b) 破坏PAM位点,例如将NGG中的一个G替换成其他碱基;c)为了筛选正确的转化子,引入一个或者消除一个酶切位点。最好选择常用的,且酶切效率较高的限制性酶切位点(EcoRI,BamHI,HindⅢ,NdeI,XbaI,XhoI),如图1E。此外,在正向和反向引物的5’末端分别添加40bp的sgRNA两侧的同源序列(表2,MIT1-repairF/ MIT1-repairR);(3)筛选引物设计。在sgRNA的两侧设计正向和反向引物,扩增出一条1kb左右的条带。两条引物与sgRNA的位置在200bp以上(见表2,MIT1-CF/ MIT1-CR)。
2.2 sgRNA克隆
用限制性核酸内切酶BsmBⅠ酶切2 μg载体pV1093,55 ℃酶切20 min后用CIP酶37 ℃处理1 h,用片段纯化试剂盒纯化酶切产物。同时用T4 Polynucleotide kinase对sgRNA的引物进行退火添加磷酸基团。最后将退火后得到的寡聚核苷酸片段与载体用T4 Ligase进行连接。
2.3 白念珠菌的转化
将克隆获得的质粒pV1093-sgRNA用KpnⅠ和SacⅠ进行酶切,并纯化回收;同时以修复模板DNA引物做PCR获得修饰模板,并纯化回收。将回收后的质粒和模板DNA用LiAc转化方法同时转化进入白念珠菌细胞。具体操作步骤为:将白念珠菌细胞在YPD液体培养基中过夜培养后,转接到5 mL新鲜的YPD液体培养基中,调整OD600为0.1,培养5 h后3000 r/min离心5 min收集细胞。用0.1 mol/L的LiAc洗涤细胞1次,再次用3000 r/min离心5 min收集细胞,最后用100 μL 的0.1 mol/L LiAc将细胞重悬,并加入10 μL提前煮沸过的10 mg/mL的Salmon Sperm DNA (ssDNA),2 μg酶切后的质粒以及2 μg的修复模板DNA模板,吹吸混匀后加入600 μL 40 % PEG/0.1 mol/L LiAc缓冲液,充分混匀。42℃热激30 min,离心弃上清后,用2 mL YPD培养基转移到试管中,200 r/min、室温(25 ℃)复苏过夜后,取1.5 mL培养物离心水洗,涂布在含有200 μg/mL NAT的YPD平板上,30℃培养2~3 d,将获得转化子在200 μg/mL NAT的YPD平板上划线,30℃培养1 d后进行菌落PCR验证。
2.4 菌落PCR验证转化子
菌落PCR主要包括两个步骤:一是处理细胞获得足够质量和数量的基因组DNA;二是PCR反应。
具体操作步骤:1、基因组DNA的制备。在冰上配制裂解体系:1/50体积Lyticase(20 Unit/μL),5X Taq Buffer,加水补充体积,并分装到PCR管或者96孔PCR板中,每个样品25 μL的体系;用扁头牙签挑取转化子,在裂解体系中混合均匀,室温放置30~60min;加入100 μL水,95℃处理5min,使蛋白失活;3000 r/min离心10 min,上清液即基因组DNA。2、PCR反应。根据所用Taq酶的使用说明进行PCR的体系的配制和程序设置。
2.5 表型实验
挑取菌株单菌落接种到3 mL YPD培养基中,30 ℃ 220 r/min 过夜培养16 h,调整OD600值为2,倍比稀释得到细胞浓度分别为2×107cells/mL、2×106cells/mL、2×105cells/mL、2×104cells/mL和2×103cells/mL。每个菌株,每个浓度吸取2.5 μL,点到YPD和含有不同药物的固体培养基上,从右到左浓度递增,在30℃培养箱培养2~4 d拍照观察结果。
3 结果
3.1 CRISPR/Cas9方法构建白念珠菌CaMIT1基因的突变株
按照Fink教授实验室构建的白念珠菌CRISPR/Cas9体系[16](见图1),我们首先构建含有CaMIT1的sgRNA的 pV1093质粒,并通过测序验证, 得到正确的pV1093- sgMIT1质粒。用KpnⅠ和SacⅠ同时对pV1093- sgMIT1质粒进行酶切,获得11 kb 和3 kb左右的两条片段 (见图2D) ,其中大片段包含有CaCas9p表达体系、sgRNA表达体系,以及NatR基因,并将通过片段两端的ENO1基因同源序列整合到基因组的ENO1基因位点。pV1093- sgMIT1质粒双酶切产物与修复模板DNA(见图2E)一起转化到白念珠菌的野生型菌株SN148,在含有200 μg/mL 的YPD固体培养基上能获得100以上的转化子。对转化子进行基因型验证(图2F、2G)。通过PCR,以野生型菌株和CaMIT1突变株的基因组DNA为模板,都能扩增出1kb左右的DNA条带(见图2F)。由于经过CRISPR编辑后的基因组序列中引入了一个XbaⅠ酶切位点,而野生型基因序列中没有,所以用XhaⅠ酶对PCR产物进行酶切后,从CaMIT1突变株基因组中扩增出的1kb PCR产物会被切成400 bp和660 bp两个小片段,而从未经过编辑的野生型菌株基因组中得到的1kb PCR产物则不会被切成两个小片段(见图2G)。在15个转化子中,1、2、3、4、5、7、8、9、12、13可能是成功编辑过的突变菌株,此酶切效率达到60%以上。由于白念珠菌是双倍体,不能被XbaⅠ酶切完全的PCR产物有可能是来自杂合子的基因组DNA,所以我们挑选被XbaⅠ酶切完全的PCR产物进行测序。包括我们获得的3个转化子mit1-1(HHCA1)、mit1-2(HHCA1)、mit1-3(HHCA1)就是从9个转化子(图2中的1-5,8,9,12,13号)中经过对它们的PCR产物进行测序获得的。测序结果表明这3个转化子中CaMIT1基因都产生了预期的突变。
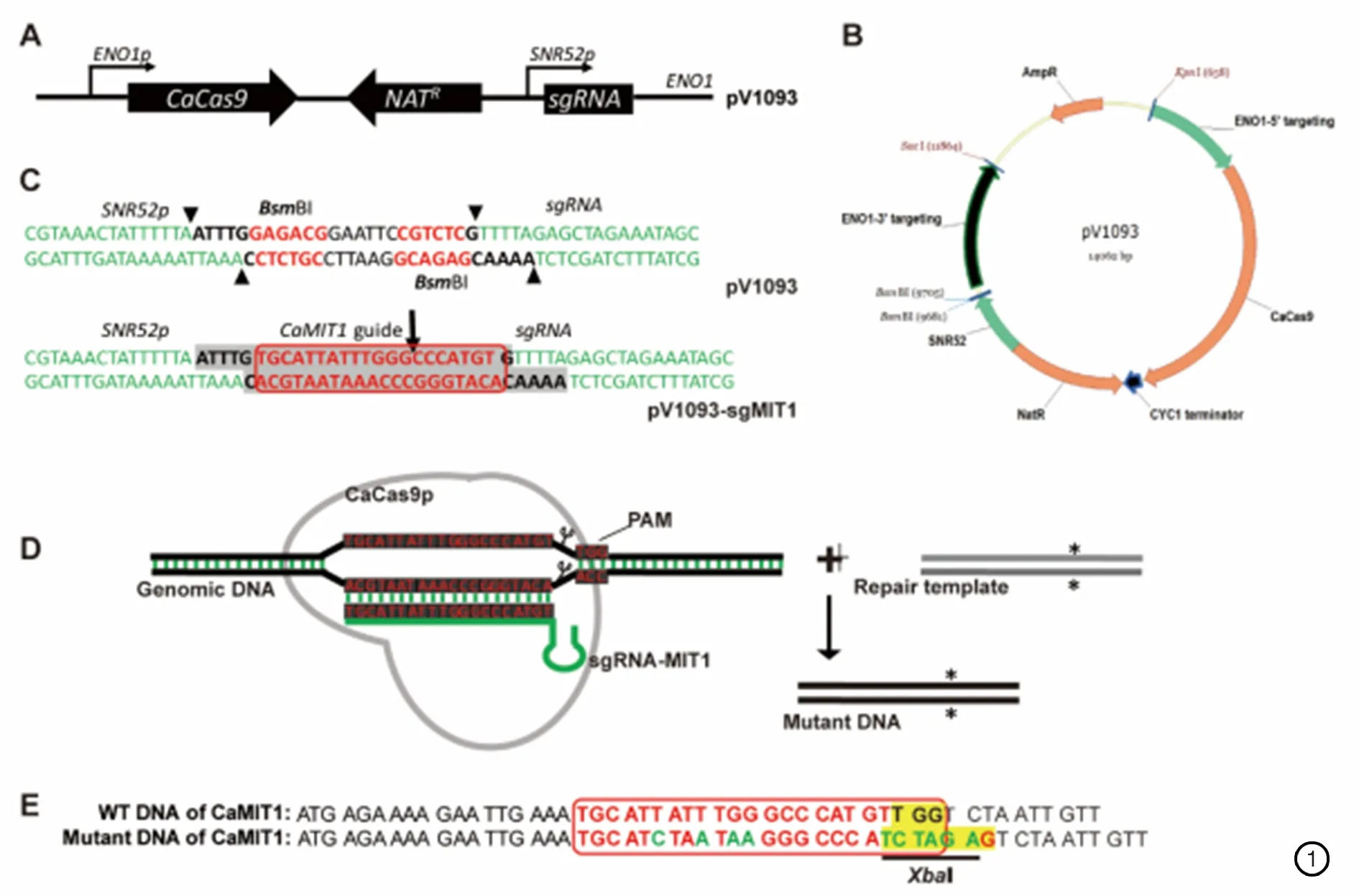
图1白念珠菌CRISPR/Cas9系统。(A) 由质粒pV1093组成的单一体系,整合到基因组上ENO1基因位置。(B) pV1093质粒图谱。CaCas9p蛋白的3’末端融合有3X SV40核定位信号和3X FLAG标签。CaCas9p的启动子是组成型表达的ENO1启动子,同时是CaCas9p整合到基因组上的整合位点。利用RNA聚合酶ⅢⅢ的启动子SNR52p来表达sgRNAs。(C) 用BsmBⅠ酶切质粒pV1093,同时将设计的导向序列(红色框架内是CaMIT1导向序列)退火,并将退火后的寡聚核苷酸(阴影部分序列) 连接到质粒上构建导向表达系统。(D) Cas9突变方法的原理图。这种系统能够在基因的特定(*,PAM位点)位点产生纯合子突变,同时使序列突变来阻止在整合后重复剪切。(E) 野生型和突变株在CaMIT1突变位点的序列。两个连续的终止密码子在CaMIT1的阅读框架(ORF)上。在PAM位点引入了XbaⅠ限制性酶切位点。
Fig.1Candida albicans CRISPR/Cas9 system. (A) The solo system consists of one plasmid, pV1093, which targets ENO1. (B) Restriction map of plasmid pV1093. The CaCAS9 gene is fused to sequences encoding the 3X SV40 nuclear localization signal and 3x FLAG tag for in-frame fusion to the 3’ end of the gene. The CaCas9p from this construct is expressed from the constitutive ENO1 promoter at the plasmid integration site. The RNA polymerase Ⅲ (Pol Ⅲ) promoter SNR52p was used to control of sgRNA expression. (C) Cloning strategy of guide sequence. oligos (shaded sequences) with desired guide sequence (CaMIT1 guide sequence in red box) are annealed, digested with BsmBI and ligated with BsmBⅠ-digested pV1093 vector. (D) Illustration of gene editing process. The CRISPR/Cas9 system creates homozygous mutations in the target gene (*, PAM site). (E) Comparison between DNA sequences ofCaMIT1 loci in the wild-type and the mutant. Two consecutive stop codons are shown within the mutated region. XbaⅠ restriction enzyme site is introduced at the PAM region.
3.2 CaMIT1突变株表型分析
在SN148遗传背景下,CaMIT1突变株对0.4 mol/L和0.6 mol/L CaCl2表现为极为敏感(如图3A)。环孢霉素A是钙离子/钙调磷酸酯酶信号途径中,钙调磷酸酯酶的抑制剂,能够抑制钙信号途径的激活[18-19]。培养基中加入50 μg/mL环孢霉素A(Cyclosporine A,CsA)不能够抑制CaMIT1突变株对钙离子的敏感表型,说明钙离子的敏感表型不是由于钙离子信号途径的激活而导致的。与Mille等人的报道一致,CaMIT1突变株对十二烷基磺酸钠(Sodium Dodecyl Sulfate, SDS),对荧光增白剂(Calcofluor White, CFW)不敏感[13];但是,CaMIT1突变株对刚果红(Congo Red, CR)表现出抗性(见图3B)。此外,我们发现CaMIT1突变株对LiCl、克霉唑(Clotrimazole, Clo)、酮康唑(Ketoconazole, KCZ)以及苯胺抗真菌药物(Anidualafungin, Anidua)表现为敏感表型(见图3A、B)。LiCl溶液通常呈微碱性,但是CaMIT1突变株对碱性环境并不敏感(见图3B)。最后,我们还发现CaMIT1突变株对DNA合成抑制剂羟基脲(Hydroxyurea, HU)敏感,但对DNA损伤剂甲磺酸甲酯(Methyl Methanesulfonate, MMS)不敏感(见图3B)。

图2白念珠菌的CRISPR是一种有效的突变系统。(A)质粒提取试剂盒提取pV1093质粒;(B) BsmBI酶切线性化质粒pV1093;(C) sgRCH1-F/R引物退火,并用T4 Polynucleotide kinase 在末端添加磷酸基团后得到RCH1的sgRNA寡聚核苷酸序列;(D)KpnⅠ和SacⅠ双酶切线性化pV1093-sgRCH1质粒;(E) PCR扩增获得修复模板DNA;(F) 转化获得转化子后,菌落PCR扩增目的片段,包含被突变位点。(G) 修复模板DNA中引入酶切位点XbaI,用限制性内切酶XbaI对PCR产物进行酶切,筛选正确的转化子
Fig.2C.albicansCRISPR is an efficient mutagenesis system. (A) Plasmid pV1093 was extracted by using plasmid extraction kit. (B) Plasmid pV1093 was linearized by digestion with BsmBI. (C) Primer of sgRCH1-F/R were annealed to obtain sgRNA oligos ofRCH1, and phosphorylated by T4 Polynucleotide kinase. (D) Plasmid pV1093-sgRCH1 was linearized by digestion withKpnIand SacI. (E) Repair DNA was obtained through PCR. (F) Colony PCR was used to amplify the flanking region including mutant site after transformation. (G) To verify correct transformants, the PCR products were subjected to digestion with the restriction enzymeXbaIwhose site was introduced during the mutagenesis

图3 CaMIT1突变株表型.
4 讨论
白念珠菌是双倍体,传统的URA-Blaster方法需要分2步才能敲除一个基因的两个等位基因,因此基因敲除过程耗时繁琐。而且,白念珠菌没有已知的有丝分裂过程,自然界不存在单倍体的形态。因此,缺少简单而快速的基因失活手段是研究白念珠菌基因功能和致病性的主要障碍。本研究通过利用Fink教授实验室构建的应用于白念珠菌的CRISPR方法成功获得了白念珠菌mit1/mit1纯合子突变菌株。通过表型实验,进一步验证了mit1/mit1突变菌株的表型与Mille等人用传统URA-Blaste方法构建的突变株的表型一致[13]。我们的工作进一步证明了这一技术的可行性。CRISPR这种高效简便的基因编辑技术的应用,无疑会加速白念珠菌基因功能的研究进程。Fink教授实验室还构建了质粒pV1200,质粒pV1200中的NatR标记含有翻转酶Flippase (NATR-FLP) ,其NATR-FLP-SNR52p-sgRNA两端含有同源序列FRT,翻转酶可以通过同源重组的原理将NATR-FLP-SNR52p-sgRNA从基因组中删除,因此NATR筛选标记又可被用于下个基因的敲除[16]。
此外,Mitchell教授实验团队已经报道了一种短暂的CRISPR基因组编辑方法[20]。这种短暂的CRISPR编辑体系,以pV1093为模板通过PCR获得CaCas9p表达体系,通过single-joint PCR获得sgRNA表达体系。以pNAT为模板,用含有同源臂的引物通过PCR扩增NAT敲除盒,或者用氨基酸合成基因做筛选遗传标记。SN148氨基酸营养缺陷菌株含有四种氨基酸合成基因缺失,包括URA3、HIS1、LEU2和ARG4。 Mitchell教授的短暂CRISPR基因组编辑方法证明sgRNA不需要整合到基因组上发挥作用。因此可以将CaCAS9整合到基因组上,利用PCR扩增获得sgRNA的表达体系和氨基酸筛选标记,通过更换sgRNA和氨基酸筛选标记获得纯合子突变菌株,以及多基因突变株。
酿酒酵母ScSUR1基因是白念珠菌CaMIT1的同源基因,和酿酒酵母ScSUR1突变株细胞一样,白念珠菌CaMIT1突变株对钙离子敏感[21]。我们的结果表明CaMIT1突变株对钙离子的敏感性不能够被CsA所抑制,说明其敏感表型与钙离子信号途径的激活无关,我们推测是由于CaMIT1缺失导致白念珠菌细胞质膜或者细胞壁存在缺陷造成的,这和CaMIT1突变株对SDS敏感这一表型结果一致[13],见图3。CFW和CR是诱发细胞壁胁迫的药物[22],它们都能够与多种多聚糖物质结合,尤其对几丁质和β-1,3-葡聚糖具有高亲和性,这两种染料都能够诱导不正常的隔膜,从而导致酵母的母细胞和子细胞在细胞分裂过程中不能够完全断裂。我们发现CaMIT1突变株对CFW不敏感,而对CR表现出耐受性(见图3)。CaMIT1突变株对这两种药物的不同表型表明这两种染料与几丁质和β(1,3)-葡聚糖的结合底物可能不同。CaMIT1缺失可能导致白念珠菌细胞壁对CR的结合度下降,而没有影响对CFW的结合度。CaMIT1突变株对破坏细胞膜通透性的抗真菌药物Clo,KCZ和Anidualafungin表现出敏感性,这与CaMIT1缺突变株对SDS敏感性表型是一致的[16]。CaMIT1突变株对Li+敏感,LiCl溶液为微碱性,但是在pH8.3以及pH10.0条件下CaMIT1突变株的生长状况和野生型细胞一致,说明CaMIT1突变株对Li+调控存在缺陷。HU和MMS都是细胞周期药物,HU是一种DNA合成抑制剂,对S期以外的细胞无影响,但是可以阻止细胞进入S期而停留在G1/S交界(看作G1期细胞)[23]。MMS是一种烷基化试剂,是DNA损伤试剂,能够使细胞停留在S期[24]。CaMIT1突变株对HU敏感,而对MMS不敏感,CaMIT1突变株应答HU的机制有待进一步研究。综上所述,CaMIT1p与白念珠菌的细胞质膜和细胞壁完整性以及钙离子的稳态相关。
