基于图卷积网络的癌症临床结果预测的半监督学习方法
2018-10-31宁世琦郭茂祖任世军
宁世琦, 郭茂祖,2, 任世军
(1 哈尔滨工业大学 计算机科学与技术学院, 哈尔滨 150001; 2 北京建筑大学 电气与信息工程学院, 北京 100044)
引言
癌症是一个复杂的世界性健康问题,因其高死亡率而受到科学家的密切关注。根据GLOBOCAN项目[1],仅在2012年,全球就有1 410万新的癌症病例(不包括皮肤癌,不包括黑素瘤),占死亡人数的14.6%。癌症类型的早期诊断和预后已成为癌症研究的必要条件。在过去的几十年里,癌症研究正日趋成熟。基因表达谱数据的利用是癌症预测研究的热点之一。基因表达谱的数据分析在很大程度上促进了癌症的诊断和治疗,准确预测癌症是医生最重要、最紧迫的任务之一。
随着高通量测序技术的快速发展,从基因组到表型,基因组各个层次的多分子水平数据,特别是基因表达谱数据越来越丰富。对于日渐增多的大型生物信息数据,机器学习方法也因此获得青睐与应用。随着近年来计算机辅助技术的飞速发展,机器学习方法在癌症诊断中的应用越来越重要。
Furey[2]提出了一种使用SVMs分析来自多个组织或细胞类型基因的卵巢、AML、结肠微阵列表达数据的方法。3种结果表明,该方法可以对组织和细胞类型进行分类。Listgarten[3]发现SVMs在乳腺癌SNPs数据集上,相比其它预测模型的表现要堪称最佳,如决策树,朴素贝叶斯等。Gevaert[4]将临床数据和微阵列数据源与贝叶斯网络相结合,提出了预测乳腺癌预后的方法。Chen[5]使用ANN构建预测模型,利用来自4家医院的样本集进行训练和预测。结果表明,4家医院的癌症患者预测结果较为准确。Kaymak[6]提出了一种乳腺癌诊断图像自动分类的方法,该方法利用反向传播神经网络(BPPN)对图像进行分类。Chougrad[7]开发了一个基于深度卷积神经网络(CNN)的计算机辅助诊断(CAD)系统,目的是帮助放射科医生将乳房X光检查分类,并获得0.99的AUC。根据文献[8],放射科医生仅获得了0.82的AUC。Xiao[9]使用集成学习策略,将多种不同机器学习模型集结融合,然后采用深度学习的方法对5个分类器的输出进行集成,在TCGA的LUAD等数据集上获得了较高的准确率以及AUC。
使用机器学习方法进行基因选择的方法也很多。Ding[10]使用MRMR来选择对白血病、结肠癌等疾病的分类至关重要的基因。Wang[11]通过filter、CFS和wrapper选择的基因,并利用这些基因构建的分类器在白血病等数据集上获得了较好的分类性能。Diaz[12]提出了一种基于随机森林的基因选择方法。
然而,这些诊断方法大多是监督学习方法。但是在生物信息学中,获取标签数据是昂贵的,现有的数据常常是不充分的。TCGA数据库已经是世界上最大的数据库之一,很多疾病仍然缺乏标记数据。例如,MESO只有86个样本,KICH也仅有89个样本。而半监督学习非常适合标签稀疏的场景。
在本文中,研究贡献有2方面。一方面,将图卷积网络(GCN)应用于基因表达谱数据,用以判断样本是否患癌。其次,研究创新性地提出了一种基于GCN的基因选择方法。在TCGA中3个数据集的实验表明,研发模型即便使用更少的样本训练,也能在分类精度和AUC上超过许多经典的机器学习方法。研究内容论述如下。
1 方法和数据
1.1 实验数据
研究利用来自TCGA的FPKM基因表达谱数据集,如LUAD、UCEC、COAD,来预测样本是否患癌。这3个数据集都是二分类数据集。实验数据集样本信息可见表1。
1.2 经典基于图的半监督学习方法
有一些经典的半监督学习方法,比如Self-training,生成模型等等。其中,Self-training是一种最简单的半监督学习方法。方法中只是对标记的数据进行培训。在每个步骤中,未标记点的一部分根据当前决策函数进行标记;然后,将监督方法重新训练,使用其自身预测作为附加的标记点。但缺点是,早期的错误可能会强化自己的错误。而生成模型认为样本适合概率模型p=(X,Y|θ)。其实现简单,但通常很难验证模型的正确性,如果生成模型是错误的,未标记的数据同样会加重自身错误。

表1 实验数据集信息
这2种方法不考虑样本之间的关系。基于图的半监督学习方法将考虑样本之间的关系。图半监督学习问题的关键在于研究做出的如下假设:
(1)相邻的点可能有相同的标签。
(2))相同结构上的点(通常称为簇)可能有相同的标签[13]。
在文献[14]中,就使用了基于图的半监督学习。该算法输出一个n维实值向量f=[flT,fuT]T=(f1,...,fl,fl+1, ...,fn)T,fi表示ithsample将为正样本的概率,通过设置输出阈值可以得到未标记样本标签。但是Kipf[15]认为该方法的假设过于严格,因为图的边不一定只编码节点相似性,且还可以包含更多的信息,因此文中放宽了一些假设。
1.3 谱图卷积
在文献[16]中,一个样本x∈N在gθ卷积核上的谱图卷积操作定义为:
gθ*x=UgθUTx
(1)

直觉上,在方程(1)中,x可以获取邻居节点的信息,因为拉普拉斯矩阵L包含图的全局信息。为了更快地计算方程(1), 文献[16]建议gθ可以由切比雪夫多项式来进行近似。通过设置一些参数,就可以归纳出如下的层传递公式:
(2)
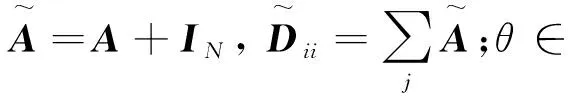
1.4 利用GCN模型判断样本是否患癌
首先,考虑一个2层的GCN来预测一个样本是否为癌症。在图(graph)中,节点xi=(xi1,xi2,...,xin)表示样本,边表示节点之间的关系。在文献[15]中,通过使用GCN表示引用网络和知识图谱中,邻接矩阵的值为{0, 1}。研究认为这种方法也可以解决邻接矩阵值为[0, 1]的连续值问题。邻接矩阵包含数据中不存在的信息,如引用网络中的文档之间的引用链接或知识图谱中的关系。与传统的标签传播思想不同,图卷积本质上不是传播标签,而是传播特征。图卷积会在不知道标签特征的情况下影响已知标签的特征节点,然后利用已知标签节点的分类器传播其属性。
(3)
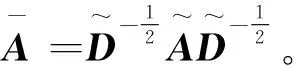
1.5 利用改进的GCN模型进行基因选择
本次研究中,还创新性地使用GCN进行基因选择,并添加一个额外的隐藏层来表示所有基因输入的基因权重。隐藏层可以通过反向传播来获取基因的权重。损失函数是交叉熵损失。选择交叉熵损失的原因是梯度下降速度快,尽可能避免梯度消失。研究运用这种方法从LUAD中选择有意义的基因。同时与一些常用的特征选择方法进行了比较。为了得到更具说服力的比较,研究选择了不同类型的特征选择方法。选取方法详情可见表2。
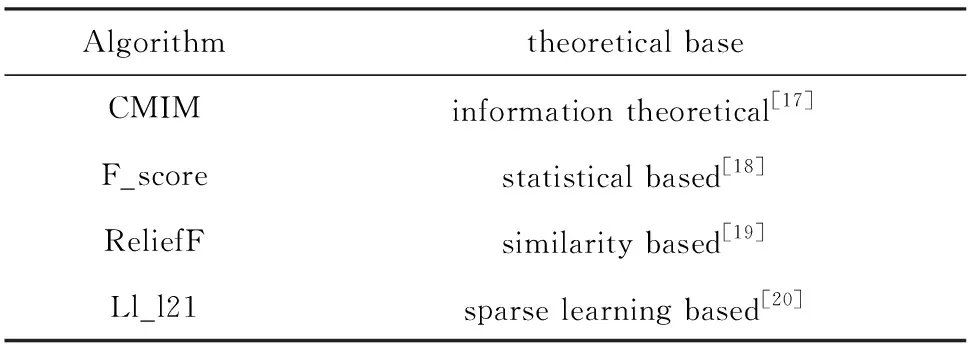
表2 基于不同理论的特征选择方法
2 实验
2.1 实验参数设置
在二分类模型中,本文的模型在3个数据集中都只训练50个样本,具体的癌症和正常的样本分布可见表3。研究则用50个样本做验证集,癌症和正常的样本比是相同的。其它的样本就用作测试。在研究选取比较的方法中,将使用100个样本来参与训练, 其中癌症和正常的样本分布可见表4。在LabelPropagation(LP)[21]中,训练样本中癌症和正常的样本分布可见表5。基因选择模型中,对于GCN,将仅会使用50个样本投入训练,样本分布参见表6。在其它比较方法中,研究使用500个样品进行训练,样本分布可见表7。
表3GCN分类模型训练集正常与癌症样本数量
Tab.3NormalandcancersamplenumberofGCNclassificationmodeltrainingset

DatasetLUADUCECCOADTumor101510Normal403540Total505050
表4KNN,决策树,朴素贝叶斯训练集正常与癌症样本数量
Tab.4NormalandcancersamplenumberofKNN,decisiontree,naivebayesiantrainingset

DatasetLUADUCECCOADTumor202020Normal808080Total100100100
表5LabelPropagation(LP)训练集中正常与癌症样本数量
Tab.5NormalandcancersamplesofLabelPropagation(LP)trainingset
DatasetLUADUCECCOADTumor101510Normal403540Total505050
表6GCN基因选择模型训练集中正常与癌症样本数量
Tab.6NormalandcancersamplesofGCNgeneselectionmodeltrainingset

DatasetLUADTumor10Normal40Total50
表7对比基因选择方法训练集中正常与癌症样本的数量
Tab.7Normalandcancersamplesnumberofgeneselectiontrainingset

DatasetLUADTumor50Normal450Total500
2.2 实验结果
在确定样本是否是癌症患者的目标预测中,给出GCN以及对比方法在3组数据集上的预测情况。准确率详见表8,AUC(Area Under Curve)参见表9。3组数据集上的ROC分别如图1~ 图3所示。
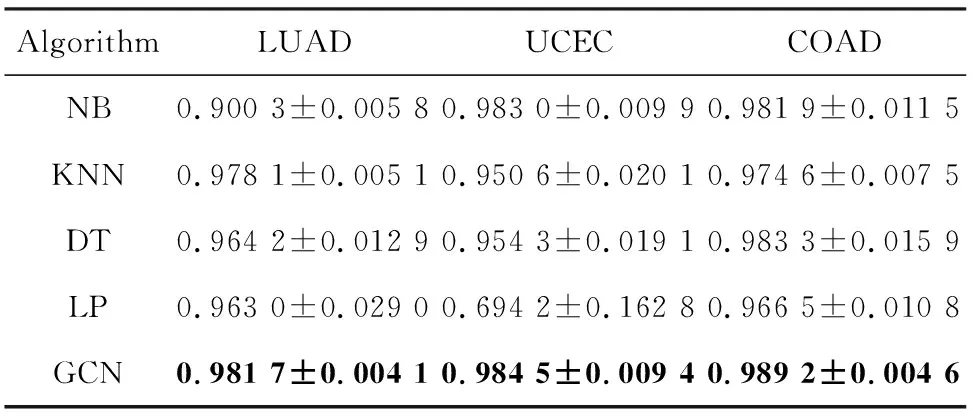
表8 3个数据集上的准确率

表9 3个数据集上的AUC
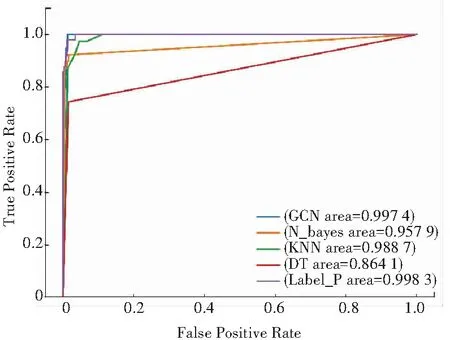
图1 LUAD ROC曲线
在基因选择的目标中,研究采用TCGA的LUAD的基因表达谱数据进行实验。关于选择出的基因的分析,本文对比了NCBI数据库。进一步获取了NCBI关于LUAD疾病的相关基因,总共1 741个相关基因,这些基因将作为结果比对的参照。

图2 UCEC ROC曲线

图3 COAD ROC曲线
这里列出了各个方法选出的top100、top200、top500基因,命中NCBI数据库的数量。各方法命中NCBI数据库的数量可见表10。

表10 各方法命中NCBI的基因数
2.3 实验结果分析
在预测样本是否患癌过程中,GCN用更少的标签数据,相比其它方法,却获得了更高的准确率,更高的AUC。究其原因在于GCN不仅考虑了样本之间的相关性,同时利用了未标记数据,通过卷积操作,获取了邻近节点的信息,最大程度保证获取信息的全面性。在基因选择目标研究中,也同样用了更少的标签数据,相比各种基于不同理论基础的方法,本文在Top100、 Top200、 Top500指标上,在NCBI中命中了更多的LUAD的相关基因。
而且,研究选取了一次实验中的Top20基因,分别是:AGER、 CLIC5、 CAV1、 CXCL14、 CLEC3B、 AGR2、 EPAS1、 SPOCK2、 EMP2、 SDPR、 SFTPA2、 RAMP3、 GAPDH、 CA9、 FCN3、MARCO、 CEACAM6、 TMEM100、 CLIC3、EDNRB,进行了生存分析,结果曲线如图4所示。在此基础上,则分析得知经过改进的GCN发现的基因,能将正常样本和癌症患者做到有效区分。并且,又随即分析发现经常出现的Top20基因中,比如CAV1、 EPAS1、 SDC1、 CLEC3B、 EDNRB等具有更小的p-values。其中,CAV1、EPAS1、SDC1等均属已被发现,且都和LUAD有重大关联的调控基因。不仅如此,还搜寻发现了一些新的可能与LUAD相关的基因,比如CLEC3B。另外,对其处理后再次发现CLEC3B在肺部表达水平排名较高,高表达水平才能维持细胞的正常代谢。由此即可推断得出,如果CLEC3B基因的变异,缺失等导致CLE3CB基因表达显著降低,将会严重影响细胞的正常功能。所以这很可能是一个与LUAD疾病相关的基因。
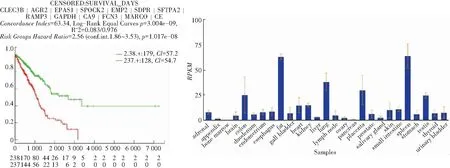
图4 LUAD生存分析曲线 图5 27个不同的正常组织样本RNA-seq表达情况
3 结束语
本文提出了一种基于图的卷积神经网络的半监督学习方法,来预测一个样本是否罹患癌症,本质上属于二分类问题。这种方法充分考虑了样本之间的相关性,通过类似于图像中的卷积方法,图中的结点(即样本)通过卷积方式,不断从相邻结点获取信息。本文在TCGA的LUAD、UCEC、COAD的基因表达谱数据集上,利用图卷积网络进行分类。相比其它经典机器学习方法,GCN使用的训练样本虽少,但是却获得了更高的准确率和AUC。
本文也改进了GCN网络结构,改进前只能用于解决分类问题,本次研究则将其用于特征选择问题,也就是选择与疾病相关的基因。研究中通过加入隐藏层,这个隐藏层的权值就是每个基因的权值。实验结果表明,改进后的GCN找出的相关基因可以很好地区分开正常和癌症样本。并且相比经典的特征选择方法,本文提出的改进后模型,可以选择出更多的疾病相关基因。同时也能探查挖掘一些未被发现的、更有潜力被选为相关基因的基因。比如CLEC3B,通过对该基因的功能分析,研究发现该基因很有可能对LUAD疾病有明显关系。
