What the future holds for the challenging hereditary spastic paraplegia?
2018-10-18JumanaBisharatKernizanCarltonWatsonKonstantinosMargetis
Jumana Bisharat-Kernizan, Carlton Watson, Konstantinos Margetis
1 Department of Surgery, Icahn School of Medicine at Mount Sinai, New York, NY, USA
2 American University of the Caribbean School of Medicine, Cupecoy, St.Maarten
3 Department of Neurosurgery, Icahn School of Medicine at Mount Sinai, New York, NY, USA
Abstract
Key words: spastic paraplegia, hereditary; hereditary sensory and motor neuropathy; spastic paraplegia genes; motor neuron disease;corticospinal tract; spasticity; review
INTRODUCTION
The aim of this review is to provide a concise snapshot of the current status of hereditary spastic paraplegia (HSP),and to bring awareness to the fact that much of the disease is still in need of elucidation.Even topics such as prevalence and global distribution of disease are in great need of study.The fascinating HSP research that has already been conducted is a mere taste of what is hopefully to come.HSP is unique in that it encompasses an enormous spectrum of pathophysiological concepts; allowing itself to be examined by curious minds of many areas of expertise.An electronic search of the Medline database for literature in English language pertinent to HSP from 1946 to 2018 was performed using the following keywords: spastic paraplegia,hereditary; hereditary sensory and motor neuropathy; spastic paraplegia genes; motor neuron disease; corticospinal tract;spasticity.
PREVALENCE
The scarcity of epidemiological studies of HSP around the globe leave much to the imagination in regards to its exact prevalence worldwide.Researchers have made strides in the direction of estimating disease prevalence and global HSP distribution, but still vast areas of the world including North America, South America, and the bulk of the African continent have limited data in regards to prevalence and remain largely unstudied.1In a recent study that looked at the prevalence of hereditary cerebellar ataxias (HCA) and hereditary spastic paraplegia based on 22 studies (12 of which included HSP data, 9 were HCA-only studies), there was an estimated prevalence within the range of 0.5–5.5/105with an average of 1.8/105(95% confidence interval (CI): 1.0–2.7) for autosomal dominant HSP (AD-HSP), and the range of 0.0–5.3/105with an average of 1.8/105(95% CI: 1.0–2.6) for autosomal recessive HSP (AR-HSP).1Several other studies, mostly limited to specific populations from regions in Europe, have total prevalences summarized in Table 1; there are apparent wide ranges in prevalence values.
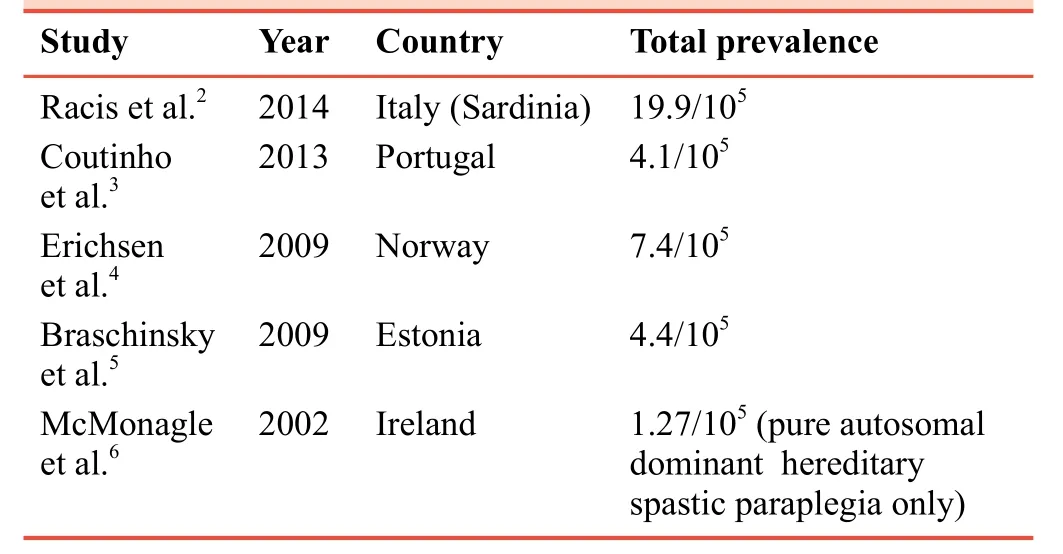
Table 1: Prevalence rates in hereditary spastic paraplegia studies
However, there are postulated sources of this discrepancy in prevalence rates.1The ideas on what could explain the variation included a multifactorial list starting first with a point that has been made by several authors.1,3,7The authors state that there is a lack of studies done in populations with no identified probands, which is quite relevant given that up to 15% of patients were identified on the basis of genetic testing alone in the study conducted by Erichsen et al.4Additionally, unstable genetic elements (i.e., migratory and founder effect, the varied rates of consanguinity in studied areas), inclusion criteria (i.e.,some studies only included only late-onset subtype patients)and under-recognition of the disease-given its wide range clinically benign and subclinical cases - all contribute to the blurring of a thorough analysis of the global prevalence of HSP.1Another challenging aspect of the epidemiology of HSP is the actual diagnosis itself, which requires exhaustive exclusion of differential diagnosesvialaboratory analysis,electromyography, nerve conduction studies, neuroimaging as well as genetic studies, which may not be as readily available in all regions globally.8
However, despite the lack of large volumes of data in this area, some trends and conclusions are evident.The AD-HSP variant, spastic paraplegia gene (SPG4), was most prevalent in all cases reviewed by Ruano et al.1X-linked and mitochondrial modes of inheritance, which have been described in several studies9-11and summarized by Fink,8do not have as much established data on gene frequencies across populations.On the other hand, another interesting conclusion is the lack of genetic diagnosis after testing, which was reported by Coutinho et al.,3in which up to 82% of cases had unidentifiable genetic culprits.In summary, although falling under the generally accepted definition of a rare genetic disease in all studies thus far, there is much regarding the prevalence and incidence of HSP that is yet to be elucidated, and the need for an organized framework for future studies is greatly needed.7
PATHOPHYSIOLOGY
An ever evolving discussion in HSP is the identification and elucidation of pathophysiologic mechanisms underlying this disease.With each gene identified, a protein product can be investigated in the hopes that a functional correlation will follow.To date, there have been numerous such studies.12-14The basic mechanistic principal is that SPG,viaa number of pathways,leads to a degeneration of neuronal axons with their terminal region being most severely affected.8,13,15The long descending motor tracts and ascending tracts of the posterior column are most affected; however, cerebellar and cortical neurons have been implicated as well.15-17Observations of terminal axonal swellings and decreased volume of spinal cord tracts in animal models and post mortem specimens have been described as evidence for this length dependent degeneration.8,18From this,it has been postulated that the HSP mutations as a group result in cellular damage that manifest most severely in the neuronal tracts with the longest axons.8,13,15
Degeneration of a portion of a cell can be accomplished in many ways and thus far, SPG have been implicated in a gambit of molecular tasks that result in this phenomenon.8,13,16Microtubule formation, vesicle trafficking, endosome formation, fatty acid metabolism, phospholipid metabolism,myelin dysformation, DNA repair and neurodevelopment are all established roles of SPG to date.8,13,18In some cases,the damaging effect may be on a supporting cell rather than the neuron itself.8These diverse cellular processes and their resultant shared clinical presentation of lower extremity spasticity are analogous to a model such as that of pneumonitis.It can be caused by a host of microbes, viruses, fungi, radiation and more; each with complex and distinct cellular targets and molecular processes, but the finding of consolidation on chest radiography and alveolar wall inflammation on microscopic examination are largely shared; of course, each with its unique standouts in addition.
The pathophysiology of SPG4 is one of the more well studied.This gene, also called SPAST, is translated into a protein product called spastin.10,13,15Spastin is characterized as having a specialized ATPase that is associated with variouscellular activities, microtubule binding regions, and several splice sites resulting in different isoforms.13,15The function of this protein is to regulate the dynamics of microtubule plus ends.Without it, uncleaved and ultra-stable microtubules can be found.13,15
There still remain many unanswered molecular questions;what mediates the fact that some HSP patients present in infancy and others as late as the 7thdecade? Why do different family members with the same mutation show such variety in clinical symptomatology? What environmental factors could also be working to drive HSP phenotypes? How much is neurodevelopment involvedversuspurely degenerative processes? The fascinating emerging science behind the gut microbiome and its involvement in neurological diseases such as Alzheimer’s disease, multiple sclerosis (MS) and Huntington’s disease may also play a role but has yet to be studied in the HSP population.19The continued efforts at studying these mechanisms are critical, because with a better understanding of each SPG pathophysiology, we can look forward to focusing research on directed treatments and cures.
cLINICAL fEATURES AND nATURAL HISTORY
There are several key features present in HSP.These features were initially used to categorize the disease into two groups,pure and complex.17,20The three symptoms, which originally made up the pure form included symmetric bilateral lower limb spasticity, urinary urgency, and decreased lower extremity vibratory sensation.20,21The complex form, on the other hand, included these three symptoms of pure HSP plus any combination of a myriad of other neurological signs and symptoms: peripheral neuropathy, epilepsy, and dementia to name a few.20-22Historically, sparing of both the bulbar and upper extremity tracts were a necessity to meet criteria for a diagnosis of HSP.20Interestingly, upper extremity and sensory involvement were described as occurring frequently in a study of pure and complex Italian HSP patients.23In addition to the neurological findings, HSP can present with a host of psychiatric comorbidities.24A study reported cases in which there was a correlation between loss of function mutation in the SPAST gene and mental health disorders that include but are not limited to severe depression, personality disorders, mania,and autism spectrum disorders.24These findings demonstrate the variety and complexity of symptomatic presentations across HSP patient populations.
More recently, with the ongoing discoveries of spastic paraplegia genes (SPG) the true complexity of the genotypephenotype relationship of HSP has been revealed and the traditional grouping into pure and complex forms is not as applicable.10,25Genes that were thought to cause pure HSP have been shown to cause complex forms as well, andvice versa.10,17Patterns of inheritance also do not follow a linear relationship with phenotype as autosomal dominant, autosomal recessive, and X-linked patterns have been shown to be implicated in either pure or complex forms of varying severity.10However, there are features that present more commonly with some mutations, for example, it has been reported that there is an association between SPG11 and a thin corpus callosum.Some SPG have even named syndromes such as SPG20, or Troyer syndrome, because the consistency in the phenotypegenotype relationship is well established.17,22To summarize these important relationships, several papers exist in which tables are presented in elegant detail about each SPG identified and the phenotypic features that are commonly observed with each one.10,17,18,20,26,27
HSP is categorized according to disease progression as well.17Clinically, HSP can have a benign, relentless, or severe course with a subsequent plateau progression pattern.8,17The average age of onset in the one study28was 30.8 years with a standard deviation of 18 and an age range of 0–73 years, while in another study23the average age of onset was 25.97 years with no reported standard deviation and with an age range of 0–64 years.It has also been described that early onset childhood cases tend to endure a less severe progression with an eventual plateau of disease progression, while late-onset cases tend to follow a more relenting course.8,29Essentially, the pure form of HSP does not affect lifespan, but can range from mild spasticity in the lower extremities to having disabling effects on mobility, quality of life and functional capacity.18,20The complex HSPs have such variability in onset time, associated neurologic symptoms and disease severity that it is difficult to comment on the natural course of these patients as a group.17,29Documented common first clinical presentations for both types are urinary urgency, or toe walking in childhood.8
In order to specifically quantify disease-related impairment,the Spastic Paraplegia Rating Scale (SPRS) was developed.28The scale is intended to monitor the progression of the disease and is especially useful for evaluating the response to treatments aimed at reducing the spasticity of the lower extremities.28The rating system is based on the accumulation of points and is easily administered as it does not require specialized equipment.Prior to this, most studies relied on the Ashworth scale for quantitative analyses of spasticity.30-32
GENETICS
A full understanding of the genetic and molecular basis of a disease is a vital component in the context of providing medical care.In the case of HSP, there is a complex and multifactorial genetic story that has only been partially detangled.In the past 10 years alone, the number of HSP genes and loci have been summarized as being 41 loci, 17 genes by Salinas et al.,1552 loci by Finsterer et al.,10greater than 80 genes and loci by Schüle et al.,2584 loci and 67 genes by Tesson et al.,18over 40 genes by Kara et al.,21and Lo Giudice et al.16reports 72 loci and 55 SPG.The true number of SPG is yet unknown,and with each study of the HSP population’s genetics, there are several novel genes described as well as those that remain undiscovered.21,25In the study of 619 HSP patients done by Schüle et al.,255 new mutations were discovered, and of the 47% of cases that were apparently sporadic, 72% did not have identifiable genetic mutations.
Thus far, HSP has been well established as having autosomal dominant, autosomal recessive, X-linked, and mitochondrial inheritance patterns.8,10,21,31There is also speculation that non-mendelian patterns of inheritance including digenic and polygenic mechanisms may also be at work.21,25Complicating things further, is the discordance in reproducibility between genetic and clinical features making it challenging to test,diagnose, categorize and anticipate the clinical course of HSP patients based on genetic analyses alone.10,18,33Even in cases where extensive molecular and genetic testing is available, the predictive value for a clinical course has a highly unreliable correlation to known mutations.21
HSP can be described as having not only genetic heterogeneity, but allelic heterogeneity, and genetic pleomorphism as well.Families with the same SPG mutation experience a wide spectrum of onset times, symptoms, and clinical course.10Meanwhile, similar phenotypic assessments have been made for a set of distinctively different gene mutations across many of the chromosomes.10Also, there are cases in which a single allele can undergo many different mutations to produce an HSP phenotype.18In SPG4 alone, there are missense, and nonsense mutations, deletions and insertions along various portions of the length of the SPAST gene.13
Some studies have described overlap between SPG and other diseases such as Chediak Higashi, amyotrophic lateral sclerosis (ALS), Charcot Marie Tooth, and Plezaues-Merbacher in a “same gene, different mutation, different disease” fashion.15,34Montecchiani et al.34reported an association between SPG11 and autosomal recessive axonal Charcot-Marie-Tooth disease.Furthermore, Orlacchio et al.35described the lack of a thin corpus callosum and the presence of amyotrophy placing emphasis on the association between SPG11 and ALS.It has been suggested that HSP lies in more of a spectrum of neurological disease rather than existing as a separate entity.This spectrum includes pathologies such as primary lateral sclerosis and spinal muscular atrophy.18,21Despite the distinct modes of HSP inheritance, Novarino et al.36suggested an HSP interactome, where the HSP genes are highly connected and link HSP to cellular transport, nucleotide metabolism and synapse/axon development.
IMAGING fINDINGS
HSP, in line with its other elusive and highly variable characteristics, has historically been indistinguishable on imaging studies.29,37The use of brain magnetic resonance imaging (MRI) is still recommended during initial work up to rule out differential diagnoses, but has also been useful for following, or staging the disease.23,29,38In many cases, standard T2 weighted MRI in patients with pure HSP has been shown to be unremarkable,while complex HSP patients have been diagnosed with brain atrophy and/or white matter changes.37-39There have been reports of spinal cord atrophy, some with a selectivity for the cervical region, and some with the diminishment of the cord in its entirety.33,40,41Interestingly, it was reported that there was insignificant correlation between degree of spinal cord atrophy and clinical symptoms in a study.40
There are some key stand outs that must be discussed in regards to HSP imaging.SPG11, the most pervasive AR-HSP documented and accounting for an estimated 15–21% of cases of AR-HSP is well established to have bilateral thinning of the corpus callosum as a consistent finding.1,39,42,43This finding of genu fiber hyperintensity on T2 weighted MRI, has been referred to as the “ears of the lynx” sign and was thought to be present with SPG11 alone, although recently, the ears of the lynx sign has been described in patients with mutations besides SPG11.43Ears of the lynx was characterized as having excellent specificity and sensitivity when comparing HSP against MS and normal control.43However, this has inherent limited utility given that most HSP patients do not carry the SPG11 mutation and in the same way, not all SPG11 mutation carriers will present with this characteristic imaging finding.A study looked at the utility of clinical presentation and neuroimaging and developed an algorithmic approach not only to determine a pure or complex HSP etiology, but to narrow down a particular gene inheritance primarily off of known genotypic-phenotypic associations.27
Some of the more promising HSP imaging data suggest that there may actually be a way to identify HSP patients of all mutant varieties, regardless of clinical expressivity.Diffusion Tensor Imaging (DTI), based on the diffusion of water through substrates is used to evaluate white matter microstructure.23,37,40Fractional anisotropy and median diffusivity are two analyses that can be evaluated through the use of DTI.23,37,40In a study of SPG4 only patients using DTI, there was significant white matter disturbance found in several parts of the brain compared to control subjects.38Also, SPG4 subjects were demonstrated to have white matter changes that preceded clinical symptoms and study authors concluded that DTI is a useful way to screen for HSP.38In a study of pure HSP patients carrying several different SPG mutations, findings of widespread white matter changes were again documented on DTI with variations in fractional anisotropy and median diffusivity compared to normal subjects and interestingly, no difference was shown between pure and complex disease forms.40While some studies reported that white matter changes are present in subclinical cases, others report that white matter changes are tightly correlated to severity of spasticity as graded by the SPRS.38,40
TREATMENTS
Synofzick and Schüle44suggested a theoretical framework for the classification of the HSP treatment strategies, dividing them into causal (targeting genetic/epigenetic factors, affected cellular pathways and neuronal systems) and symptomatic treatments (targeting symptoms).With no curative medications or procedures known to date, current HSP treatments are aimed at symptomatic relief.8,17,20,31With the breadth of symptoms experienced, lower extremity spasticity is the most common and thus often the first target for ameliorating efforts.26In general, these treatments are a combination of pharmacologic, physiotherapeutic, and device-based (orthotics); with an emphasis on maintaining cardiovascular fitness and functional capabilities.20,45
The lower extremity spasticity that HSP patients experience may have largely unknown and multifactorial pathophysiology, but the pharmacologic targets are shared with spasmolytic treatments used, for example, in spinal cord injury and MS.46Baclofen, which is a spasmolytic drug commonly used in HSP, is a centrally acting GABA-B agonist, works to heighten the descending inhibitory interneuron signal by way of Ca2+channel modulation.31,46,47Baclofen is administered either orally orviaan intrathecal route, and the latter has been demonstrated in several studies to provide patient satisfaction and improvement of spasticity.31,46-51The blood-brain barrier creates a limitation to oral administration, while the intrathecal route is complicated by pump malfunctions, infections, and seromas,etc.Both routes of administration carry the risk of withdrawal, and side effects including: drowsiness (more so when given orally) and the potential of serious toxicity when overdosed.31,46A special attention must be paid to the gradual dosing of intrathecal baclofen, because the spasmolytic effect may be so great it can diminish the ability of the patient to maintain upright posture by swinging the balance from spastic muscles to overt weakness.31,52Typically, a trial intrathecal dose in initiated with gradual titration upwards until a good balance is achieved.31,52Tizanidine, an alpha-2-agonist, and Botulinum toxin intramuscular injections have also been used to treat spastic lower extremities in HSP.46,53There were no specific studies of Diazepam, Dantrolene, Clonazepam, or Gabapentin in HSP in the literature, but these are commonly used in other disorders with spasticity symptoms.46
Other studies in HSP treatments while promising, are not as applicable to the HSP population as a whole, given their specific targets.SPG5, a variant of HSP is known to have defects in the cholesterol and phospholipid metabolism pathway therefore; cholesterol-lowering drugs such as ezetimibe,simvastatin, and chenodeoxycholic acid have been used as treatment.54,55Dalfampridine, a K+channel modulator that improves action potential conduction that is used in demyelinating conditions to help with spasticity has been tried in patients with several different SPGs.55,56Tubulin-binding drugs including Taxol and Vinblastine were given to SPG4 patients whose mutation is related to deficient microtubule stability and slowed peroxisome transport.57In a cell model,microtubule-targeted therapy has shown a reduction in axonal swelling, and may ultimately improve locomotor activity.55On a less molecular note, trials of robot-assisted therapy and hydrotherapy have also been described in HSP patients; all with mixed results.58,59With spasticity often being the only shared symptom between different SPG carriers, it is likely that future treatments will include physical therapy and spasmolytic medications.
CONCLUSION
There are few things we know about HSP, many things we know that we do not know and given the heterogeneity of the disease there are likely many things we do not know that we do not know.This modified quote by Donald Rumsfeld represents the current state of HSP.Much has been discovered (Figure 1),and yet there is much to be learned in this multifaceted disease.To date, research is ongoing in regards to the prevalence of disease across the world; however, an organized framework for future studies would be useful in extracting such data with the hope for a greater understanding of the many aspects of HSP.Thus, the aim of this review is to build awareness of HSP and encourage future studies in hopes that efforts be put towards clinical application with the goal of improving the quality of life for those affected.
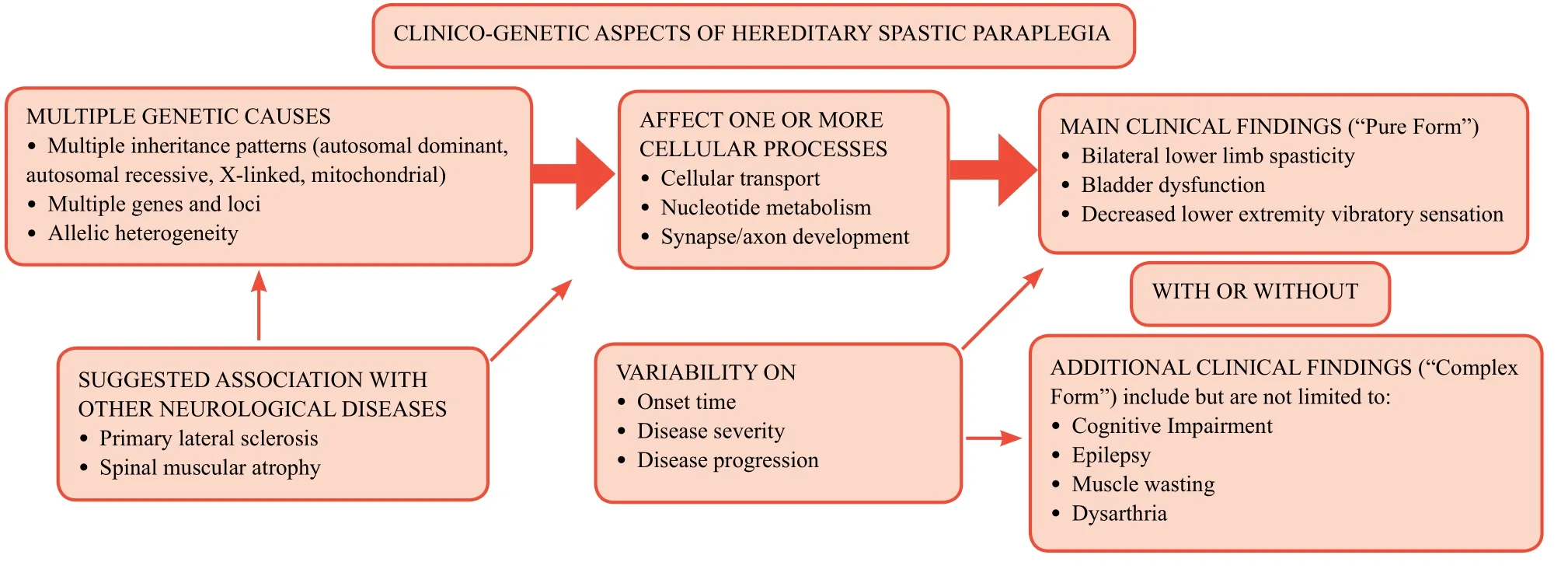
Figure 1: A schematic providing a simplified overview of the clinico-genetic aspects of hereditary spastic paraplegia.
Author contributions
Manuscript writing: JBK; manuscript editing: CW; project supervisation: KM.
Conflicts of interest
None declared.
Financial support
None.
Copyright license agreement
The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check
Checked twice by iThenticate.
Peer review
Externally peer reviewed.
Open access statement
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
杂志排行

Clinical Trials in Degenerative Diseases的其它文章
- Systematic review on effectiveness of theory-based intervention on self-care behaviors among patients with type 2 diabetes
- High focused Evaluation of Atherosclerotic risk profile in Retinal Thrombosis: Vascular events Incidence, Sex involvement and Interventional outcomes assessed by Ophthalmologists and internists Network – HEART VISION study protocol
- Accurate identification of potential critical coronary lesions for the reduction of risk of cardiovascular events: study protocol for a randomized, open-label,active-controlled multi-center trial
- Multi-component botanical drugs for degenerative diseases
- Using robotic-assisted technology to improve lower-limb function in people with stroke