绣球藤叶片转录组分析及SSR引物开发
2018-10-16陶仕珍孙正海
陶仕珍,田 斌,孙正海,张 睿
(1.云南林业职业技术学院,云南昆明650224;2.西南林业大学,云南昆明650224)
转录组是指特定生物体在某种状态下所有基因转录产物的总和。目前,以Illumina、454和ABI为代表的第2代测序技术的迅速发展及随之产生的诸如SOAP-denov等新的分析方法使人们能更全面快速地了解真核生物复杂的转录组[1-2],更为全基因组测序尚未完成(无参考基因组)的生物提供了组学研究的新途径[3]。
通过转录组测序分析能有效挖掘植物不同组织、不同生长发育时期及在不同环境下的基因表达差异[4]。另外,转录组测序对新基因的深度发掘、基因家族鉴定、代谢途径确定等科学问题提供了新的解决途径[5]。近年来,由于转录组测序成本的降低,通过转录组数据开发适用于动植物育种、濒危物种保护、群体遗传学等领域的分子标记也已获得了很好的效果[6]。
铁线莲属(Clematis)是毛茛科(Ranunculaceae Jussieu)的一个世界性分布的大属,共约有350余种[7]。我国是铁线莲属物种多样性最高的国家之一,约有147种,其中93种为我国特有[8]。这个属的植物多为大型攀援草本,并且有很多物种如转子莲(C.patens)、毛茛铁线莲(C.ranunculoides)、长瓣铁线莲(C.macropetala)等花大而艳丽,具有很高的园艺价值。此外这个属的部分植物如绣球藤(C.montana)、威灵仙(C.chinensis)、小木通(C.armandii)等还有很高的药用价值。本研究所涉及的物种绣球藤是铁线莲属绣球藤组分布较广泛的一个物种,自然分布于我国西南以及喜马拉雅地区,具有良好的药用价值和观赏价值[9]。
本研究拟利用Illumina测序平台对绣球藤叶片的转录组进行测序,对其测序组装得到的unigene进行分类统计、功能注释以及代谢通路分析,并得到一批简单重复序列(simple sequence repeat,简称SSR)位点,为今后对铁线莲属植物资源的深入研究和有效利用奠定基础。
1 研究方法
1.1 总RNA的提取和测序
用十六烷基三甲基溴化铵(hexadecyltrimethylammonium bromide,简称CTAB)法[10]提取采自云南省昆明市的绣球藤新鲜叶片的总RNA。RNA的质量和浓度用琼脂糖凝胶电泳及Nanodrop 2000进行检测和测定。随后,分别采用OligodT磁珠试剂盒从质量、浓度合格的总RNA中分离纯化出mRNA。加入缓冲液将纯化的mRNA打断成短片段,并以mRNA为模板,用六碱基随机引物合成单链cDNA。合成单链cDNA后,加入缓冲液、dNTPs和DNA聚合酶Ⅰ合成双链cDNA,并利用AMPure XP beads纯化。纯化的双链cDNA经过末端修复、加多聚腺嘌呤尾(ploy A),连接测序接头,选择片段大小以及PCR富集后成为最终的cDNA文库。最后将获得的cDNA文库采用高通量测序平台Illumina HiSeqTM2500进行转录组测序。
1.2 无参拼接及聚类
获得转录组原始测序数据后,采用自行开发的perl编程脚本对原始测序数据进行质量控制和数据过滤。质量控制及数据过滤过程包括4个步骤:(1)去除包含测序接头的序列;(2)去除N(N表示无法确定的碱基信息)的比例大于10%的序列;(3)去除低质量序列(质量值小于10的碱基数占整个序列的50%以上的序列,其中质量值按照公式Q=-10lg e计算,式中:Q为质量值;e为测序错误率(不正确的碱基识别率);(4)去除冗余序列。数据过滤后,将所有过滤序列混合在一起,采用Trinity软件[2]进行拼接。该软件拼接过程包括3个步骤:虫、蛹和蝴蝶,经过这3个拼接步骤后,得到包含大量简并转录本和可变剪接形式的完整序列信息。最后,用CD -HIT软件[3](http://www.bioinformatics.org/cd -hit/)对拼接后的序列进行聚类以及冗余序列的去除,聚类后得到的转录本为unigene。
1.3 同源搜索及功能注释
采用NCBI的本地Blast对聚类后得到的转录本进行数据库同源搜索,进行相关功能注释,搜索的期望值(E值)设定为 10-5。用于搜索的数据库有 NR(the NCBI nonredundant)数据库,Swiss-Prot(swissprot protein database)数据库和KOG(eukaryotic ortholog groups)数据库。此外,采用Blast2go 软件[11]对 unigene进行基因本体论(gene ontology,简称 GO)注释,并采用 WEGO 网站[12](http://wego.genomics.org.cn/cgi-bin/wego/index.pl)作 GO 功能分类统计,从宏观上认识绣球藤的基因功能分布特征。
1.4 SSR位点搜索和引物设计
采用 MISA(MIcroSAtellite identification tool,http://pgrc.ipk-gatersleben.de/misa/)对 unigene进行 SSR位点查找。查找的原则为所有SSR位点为一至六核苷酸的碱基重复类型,其中单核苷酸重复次数最少为10次,二核苷酸重复次数至少为6次,三核苷酸、四核苷酸、五核苷酸和六核苷酸重复次数至少为5次。
2 结果与分析
2.1 绣球藤转录组数据的组装结果
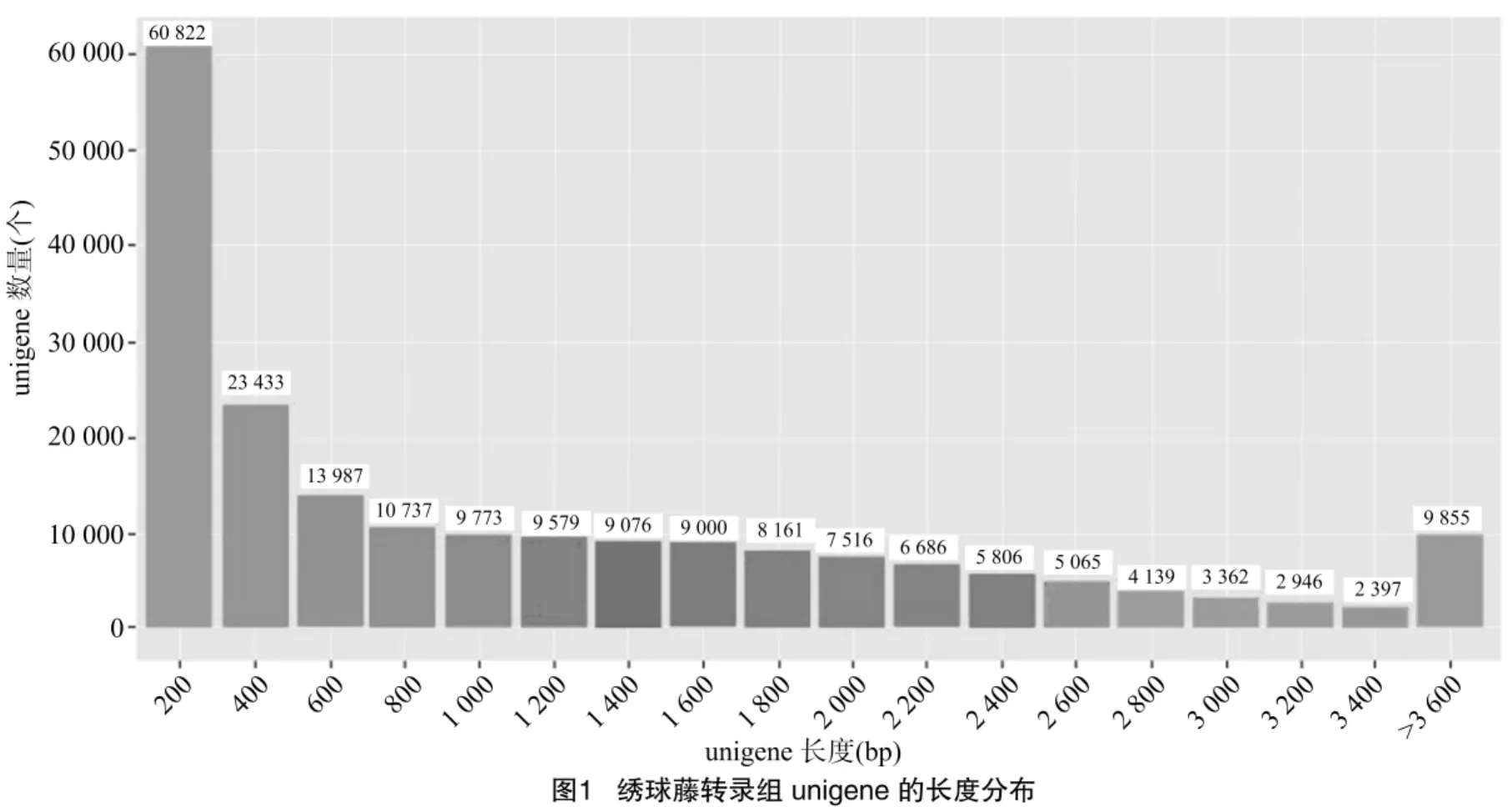
采用Illumina HiSeqTM2500高通量测序平台对绣球藤叶片转录组进行测序,共得到原始reads 92 042 086个,经过滤后的reads数为80 406 986个,过滤后用于组装的数据量约为8 GB。随后,通过Trinity软件进行无参拼接并用CD-HIT软件进行聚类以及冗余序列的去除。经过拼接及聚类,最终得到的绣球藤转录组unigene的长度分布情况(图1)。总共获得202 340个unigene,长度分布于201~19 415 bp之间,平均长度为642 bp,N50(按照长度将拼接转录本从大到小排序,累加转录本的长度为总长50%的拼接转录本长度)为646 bp。
2.2 unigene的功能注释、分类和代谢途径分析
2.2.1 数据库同源比对及注释 对聚类后得到的转录本进行NR数据库、Swiss-Prot数据库和KOG数据库同源比对及注释。在E值为10-5的条件下,共95 586个unigene与3个数据库序列具有同源性,占总数量的47.24%。与NR数据库序列有同源性的 unigene最多,共 94 904个,占总数的46.90%,其中64 655个(31.95%)的 unigene与 NR 数据库有较高同源性(E≤ e-50)。其次是KOG数据库,共24 774个unigene分类至24个功能,占总数的12.24%。在这些功能分类中,只能大致预测功能的 unigene数量最多,3 957个unigene,占15.97%,其次是翻译后修饰、蛋白质转运和分子伴侣功能,2 042 个 unigene,占 8.24%;1 765(7.12%)个unigene具有翻译,核糖体结构的功能;1 388个 unigene(5.61%)具有糖类运输及代谢功能;1 025个 unigene(4.14%)具有氨基酸转运及代谢功能,最少的功能分类为细胞运动性功能,仅0.1%unigene具有该功能。3个数据库中,比对后同源性最低的为Swiss-Prot数据库,仅65 288个unigene与该数据库具有同源性,E值大多分布在e-150~e-5之间。
2.2.2 GO功能分类 GO是一套国际标准化的基因功能描述的分类系统。根据功能,GO可分为三大类:细胞组分(cellular component)、分子功能(molecular function)和生物过程(biological process),分别用来描述基因编码的产物所处的细胞环境、所具有的分子功能及所参与的生物过程。采用Blast2go软件对unigene进行GO注释,并采用WEGO网站作GO功能分类统计,可得到图2结果。所有unigene中,可将64 542个unigene划分至三大类38个功能亚类,其中22 166个unigene具有细胞组分功能,27 092个unigene具有分子功能,15 288个unigene具有生物过程功能(单个unigene可同时具有多个功能)。从三大类来看,细胞组分大类中,具有细胞和细胞组成功能的unigene最多;分子功能大类中,具有绑定、催化活性功能的unigene最多;而生物过程大类下,具有细胞过程和代谢过程的unigene数量最多。通过基因功能注释过程,可从宏观上认识绣球藤的基因功能分布特征,为后期的研究奠定基础。
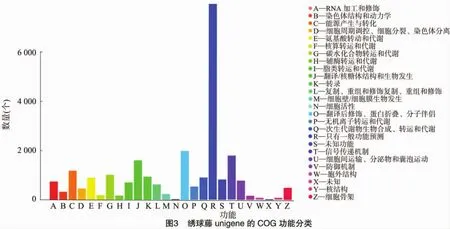
2.2.3 unigene的COG功能分类 对绣球藤unigene的蛋白质直系同源数据库(cluster of orthologous groups,简称COG)功能分类结果表明,绣球藤注释到COG的unigene根据功能大致可分为26类,并对每类的unigene数量进行统计分析,结果见图3。其中,一般功能预测类基因最多(7 978个);其次是翻译后修饰、蛋白质折叠和分子伴侣类基因(1 986个)及信号转导机制(1 800个);而细胞活性(8个)和未知功能(1个)的基因较少。

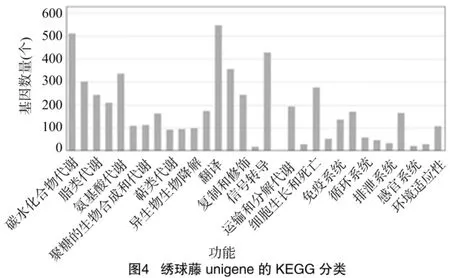
2.2.4 unigene的KEGG分析 笔者所在实验室对KEGG注释结果作了分类统计,根据参与的KEGG代谢通路,将基因分为5个分支。其中,细胞过程552个,环境信息处理449个,遗传信息处理1 330个,代谢2 282个,有机系统772个。由图4可知,在所有的注释中数量最多的3个分类为翻译550个,碳水化合物代谢(carbohydrate metabolism)515个,信号转导(signal transduction)360个(图4)。这些注释为后续次生代谢物的合成和代谢提供了很多有价值的信息。
2.2.5 SSR分析 对组装后的绣球藤202 340个unigene进行SSR位点搜索,共检测到10 255个SSR位点。通过位点搜索,没有发现六核苷酸重复类型。其中,单核苷酸重复为5 224个,占50.94%;二核苷酸重复为2 590个,占25.26%;三核苷酸重复为1 952个,占19.03%;四核苷酸、五核苷酸重复所占比例较低,分别仅有58、13个。另外在检测到的SSR位点中,复合SSR出现了418个。由于单核苷酸SSR位点在实际运用中较难统计,因此在基序统计中去除了单核苷酸重复的位点,结果显示,二碱基重复出现频率最高的3类基序为GA(540个)、AG(485个)、TC(396个);三碱基重复出现频率最高的3类基序为GAA(115个)、AGA(111个)、AAG(110个)。上述SSR特征分析有助于开展绣球藤及其铁线莲属植物的微卫星序列差异分析、分子标记开发和高密度遗传图谱构建的研究。
3 结论
利用Illumina测序平台对绣球藤叶片的转录组进行测序和分析,首次揭示了绣球藤转录组的基本表达模式,初步获得了一批在绣球藤叶片中表达的基因序列信息,共获得202 340条质量较高的unigene,其中得到注释的有95 586条,表明铁线莲属植物存在大量的未知功能基因,为深入开展药用植物生物活性成分的合成和鉴定提供了丰富的数据资料。在利用现代分子生物学技术提高天然药用植物有效成分产量以及相关药物研发和生产方面,具有重要的理论和应用价值。另外,笔者所在实验室发现的绣球藤SSR位点能开发出大量适用于绣球藤的分子标记,为今后分子标记辅助育种及群体遗传学研究提供了第一手研究资料,并且结合这些unigene基因信息和SSR位点能对铁线莲属植物的系统进化研究提供帮助。
