基于凝胶渗透色谱及液相色谱串联质谱测定油脂性食品中的维生素A、D、E
2018-10-13李珉张莉余婷婷范志勇
李珉,张莉,余婷婷,范志勇
(湖北省食品质量安全监督检验研究院,湖北省食品质量安全检测工程技术研究中心,湖北武汉 430000)
维生素 A、D、E极性较弱不溶于水,易溶于有机溶剂,属于脂溶性维生素(Fat Soluble Vitamins),是人体必需的营养素,在食品及保健食品领域的研究和应用非常广泛。食用油脂中天然脂溶性维生素含量高低是评价油品品质和营养价值的重要参数,维生素A、D作为营养强化剂用于食用油中可提高其营养价值,维生素E在油脂加工中作为天然抗氧化剂防止油脂氧化,保障油品品质。保健食品中以油脂为载体加入维生素A、D、E用以预防夜盲症、佝偻病和抗衰老等[1]。然而,脂溶性维生素在高温、高湿、光照、和极端pH值条件下易发生转化,若在食品加工储存环节流失严重,则无法达到预期的营养强化目的。同时脂溶性维生素的过量摄入还存在着蓄积中毒的风险,如维生素A摄入过量可能导致胎儿畸形、维生素D摄入过量可能引起低热惊厥、维生素E摄入过量可能导致出血倾向并影响机体免疫功能[2]。因此,实现快速准确地定性定量分析油脂性食品中的维生素 A、D、E能够为油脂性食品质量安全监管及油脂抗氧化新工艺研究提供技术支持[3~5]。我国现行的检测方法标准[6~8],国内[9~12]和国外[13~16]的研究表明,目前应用最为广泛的脂溶性维生素A、D、E的测定方法为皂化后液液萃取-液相色谱法,其中仅维生素D有现行国家标准方法可通过皂化后液液萃取后由质谱检测器检测结合内标法定。皂化后液液萃取法优点,一是样品净化较为彻底,二是统一了脂溶性维生素的检测形式;但操作步骤繁杂、耗时长,无法实现快速批量检测,同时过多的前处理操作环节极易影响结果的精密度。凝胶渗透色谱(Gel Permeation Chromatography,GPC)可用于对复杂基质样品中脂肪、色素和蛋白质等大分子干扰物质的处理净化,能够显著提高前处理的效率和结果的精密度[17]。质谱检测作为高灵敏度的定量检测技术,十分适用于油脂性食品中脂溶性维生素的精准定性和定量[18~22]。
本实验采用 GPC技术对油脂性食品进行自动化的前处理,采用液相色谱串联质谱同时测定脂溶性维生素A、D、E,从而显著提高油脂性食品中脂溶性维生素检测方法的分析效率。
1 材料与方法
1.1 材料、试剂与仪器设备
乙酸乙酯(色谱纯),Merck公司;甲醇(色谱纯)、甲酸(优级纯)、乙酸铵(优级纯)、视黄醇标准样品(纯度95%),SIGMA-ALDRICH公司;视黄醇醋酸酯标准样品(纯度99.9%),Supelco公司;DL-α-生育酚标准样品(纯度99.0%),Dr.Ehrenstorfer GmbH公司;维生素D2标准样品(纯度99.1%),Dr.Ehrenstorfer GmbH公司;维生素 D3标准样品(纯度 98.9%),Dr.Ehrenstorfer GmbH公司。
MS205DU电子天平,梅特勒-托利多;Elmasonic S180H超声仪,Elma Hans Schmidbaner GmbH &Co.KG公司;Preplinc GPC+Accu Vap凝胶净化渗透色谱仪,J2 Scientific公司;配备紫外检测器、分离色谱柱PT785 Bio-BeadsS-X3(填料质量50 g)、环己烷(色谱纯),Merck公司;TSQ Quantum Access Max液相色谱串联质谱联用仪,Thermo公司;配有电喷雾电离源(ESI),Thermo Xcalibur质谱工作站;Milli-Q Reference超纯水器,美国Millipore公司。
1.2 实验方法
1.2.1 配制标准溶液
分别准确称量视黄醇、视黄醇醋酸酯、DL-α-生育酚、维生素D2、维生素D3标准样品5 mg、5 mg、10 mg、10 mg、100 mg至10 mL棕色容量瓶中,精确至0.1 mg,加甲醇超声溶解后定容至刻度,作为标准储备溶液,于-4 ℃避光储存。
1.2.2 仪器条件
1.2.2.1 凝胶色谱条件
分离色谱柱:PT785 Bio-BeadsS-X3(填料质量50 g);流动相:环己烷-乙酸乙酯(5:5,V/V)溶液,流速:5 mL/min,进样量:5 mL;洗脱时间:(0~15)min、收集流出液时间:(15~35)min、清洗时间:(35~45)min。
1.2.2.2 色谱条件
色谱柱:Thermo Hypersil GOLD(2.1 mm×100 mm,粒径 3 μm);柱温:35 ℃;进样体积:10 μL;流速:0.4 mL/min;流动相:A:甲醇、B:0.1%甲酸5 mmol/L乙酸铵溶液,梯度洗脱程序:(0~1)min,50%A;(1~3)min,50%~95%A;(3~9)min,95%A;(9~10)min,95%~50%A;(10~11)min,50%A。
1.2.2.3 质谱条件

图1 各脂溶性维生素MRM色谱图Fig.1 Multiple reaction monitoring chromatograms of fat soluble vitamins
离子源:电喷雾离子源(ESI);扫描方式:正离子模式;监测方式:多反应监测(MRM);喷雾电压:3800 V;雾化器温度:200 ℃;鞘气压力:40 psi;辅助气:15 psi;毛细管温度:350 ℃。
定性离子、定量离子及对应的碰撞能量、喷雾电压参数见表1。图1为各脂溶性维生素MRM色谱图,其中视黄醇出峰时间为4.00 min,视黄醇醋酸酯出峰时间为4.48 min,维生素D2出峰时间为5.92 min,维生素D3出峰时间为6.00 min,维生素E出峰时间为6.81 min。
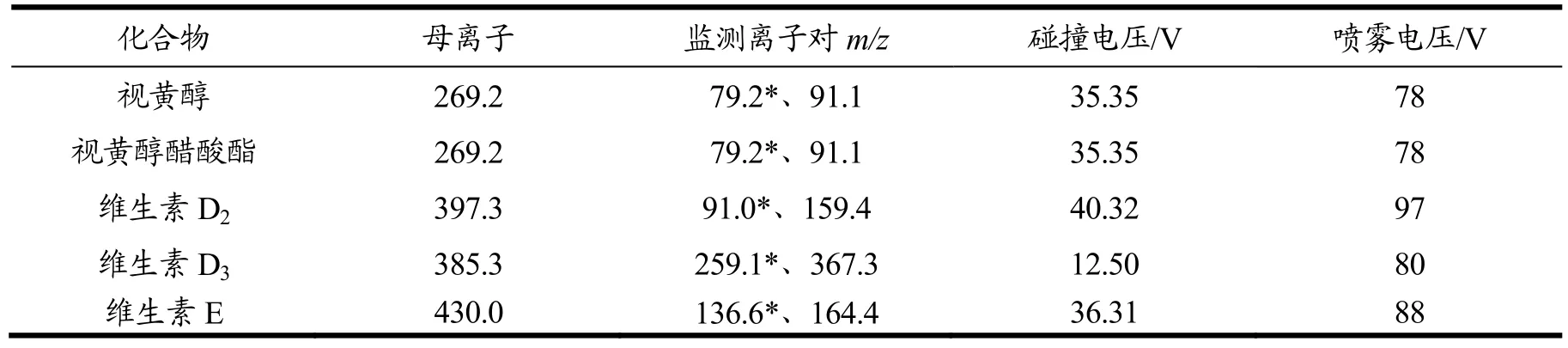
表1 脂溶性维生素的质谱分析参数Table 1 MS/MS parameters for fat soluble vitamins
1.2.3 样品处理
油脂性食品:称取0.3~0.5 g试样于10 mL刻度离心管中,在避光条件下,加入环己烷-乙酸乙酯(5:5,V/V)溶液定容至10 mL刻度线,涡旋震荡2 min,提取液用0.22 μm有机滤膜过滤后按照1.2.2.1项方法用凝胶色谱净化,流出洗脱液浓缩定容到1 mL待分析。
脂溶性维生素混和标准使用溶液:吸取标准储备液各200 μL于10 mL棕色容量瓶中,在避光条件下混匀后用环己烷-乙酸乙酯(5:5,V/V)溶液定容至刻度,提取液用0.22 μm有机滤膜过滤后按照1.2.2.1项方法用凝胶色谱净化,流出洗脱液浓缩定容1 mL待分析。
2 结果与讨论
2.1 GPC分离条件的确定
油脂样品主要由高级脂肪酸的甘油酯组成,其分子量通常在 800以上,而脂溶性维生素的分子量在250~450之间。GPC技术是基于空间排阻的原理根据分子量大小的不同进行分离,因此本实验采用GPC为净化手段。可能对分离效果产生影响的主要因素包括色谱柱类型、柱容量、流动相流速、流动相组成和比例。商品化的凝胶渗透色谱柱通常规定了允许使用的流动相组成和比例,故本实验主要考虑的条件优化因素为色谱柱类型、色谱柱容量和流动相流速。
理论上,色谱柱容量越大、流动相流速越慢越能够改善混合物的分离度,但实际操作时还要考虑检测工作的时效性,因此通过交互设计,对两种不同长度的色谱柱在三种流速下分别进行试验,结果如下:
(1)以环己烷-乙酸乙酯(5:5,V/V)溶液作为流动相,选择两种常用长度的色谱柱,分离色谱柱1:Express GPC cleanup column(填料质量20 g)、分离色谱柱2:PT785 Bio-BeadsS-X3(填料质量50 g),分别比较流速1(5 mL/min)、流速2(3 mL/min)、流速3(2 mL/min)三种不同流速下的脂溶性维生素与大豆油的分离度,在可接受的分离度下比较总分析时间,选择合适分离度条件下分析时间最短的条件作为最佳方案。当分离度为0.8时,相邻两组分可达到95%的分离度,完全满足净化油脂的需要,本实验拟将分离度达到0.8作为可以接受的分离度。表2为分离度与总分析时间情况,综合考虑分离度和总分析时间指标,试验1和试验2的分离度小于0.8,无法满足净化油脂的需求,试验3、试验4、试验5、试验6的分离度均大于0.8,且试验4所用分析时间最短,故选择试验4为以大豆油为基体分离脂溶性维生素的最佳分离方案。
分离度计算公式:R=2(t2-t1)/(W1+W2)
注:t2:相邻两峰中后一色谱峰的保留时间;t1:相邻两峰中前一色谱峰的保留时间;W1、W2:此相邻两峰的峰宽。

图2 最佳分离条件下脂溶性维生素混合标准样品和不同油脂样品的GPC图谱Fig.2 GPC chromatograms of fat soluble vitamins mixed standard samples and different oil samples at the optimal separation conditions
(2)采用以上最佳分离条件对其他油脂样品如大豆油、动物油(黄油)、维生素 E软胶囊、鱼肝油软胶囊进行分离度的测试,通过试验由图2可见,脂溶性维生素混合标准物质在15 min后流出,而大豆油、动物油(黄油)、维生素 E软胶囊、鱼肝油软胶囊中的油脂成分均在15 min前出峰流出,二者分离度均在0.8以上,结果表明该条件能够将其它品种油脂样品和脂溶性维生素进行很好地分离,满足实验需求,故选择15 min~35 min收集流出液。

表2 分离度与总分析时间Table 2 Resolution and total analysis time
2.2 色谱质谱条件的优化
2.2.1 流动相的优化
分别选择甲醇-甲酸水和乙腈-甲酸水作为流动相,试验表明:当以乙腈作为流动相时,其洗脱能力较强,视黄醇和视黄醇醋酸酯在液相部分难以分离;二者母离子和碎片离子信息完全一致,在质谱部分也无法分离;同时发现在乙腈条件下各标准样品的离子化程度均受到一定程度的抑制,且维生素 D3无法检测,以甲醇作为流动相时的检测灵敏度提高2个数量级,且所有脂溶性维生素均可检测,故选择以甲醇作为流动相。本实验为正离子扫描模式,尝试在流动相中加入甲酸、乙酸铵以提高离子化效率及改善峰型,试验表明,0.1%的甲酸、5 mmol/L的乙酸铵体系能提供最佳离子化条件。
2.2.2 离子源选择
电喷雾离子源(ESI)是最常用的液相离子源,适用于极性较强的化合物,可用于热不稳定化合物的分析大气压化学电离源(APCI)适用于中等极性或弱极性的小分子量化合物,尤其是含杂原子的化合物,不适合热不稳定或在溶液中容易电离的化合物。虽然ESI源是常用的离子源,但由于脂溶性维生素极性较弱,从原理上可能APCI源具有更好的电离效果,故对两种离子源进行对比试验,结果表明APCI源在分析维生素D2和D3时比ESI源具有更高的灵敏度,但对维生素A和维生素E的分析不如ESI源,考虑到ESI源的通用性,本实验选择ESI源。
2.2.3 母离子确定
视黄醇是一个具有脂环的不饱和一元醇,其分子量为286.45,正离子扫描下理论分子离子峰应为287,在研究质谱方法的过程中,未找到287的母离子。根据经验,脂肪族醇、胺、腈类及多支链化合物容易裂解,分子离子峰通常很弱或不出现,因此判断视黄醇在分析过程中可能发生了源内裂解,分析其分子结构,其容易发生脱氢重排脱去一分子水,形成质荷比为269的脱水峰,经试验,证实了这一判断,同时发现视黄醇醋酸酯也发生了同样的反应。
2.3 方法学实验
2.3.1 方法检出限和线性范围取浓度分别为5、20、50、100、200 μg/L的系列标准溶液,以峰面积A对相应的质量浓度C进行线性回归,结果表明:视黄醇、视黄醇醋酸酯、维生素D2、维生素D3、维生素E在5~200 μg/L的浓度范围内均
具有良好的线性关系,相关系数r均在0.999以上。按3倍信噪比计算得到样品中视黄醇、视黄醇醋酸酯、维生素D2、维生素D3、维生素E的检出限分别为2
μg/kg、1 μg/kg、5 μg/kg、2 μg/kg、10 μg/kg。
2.3.2 方法回收率
在不含有脂溶性维生素的大豆油基质中添加脂溶性维生素混合标准样品溶液,使大豆油中各脂溶性维生素含量为10.0、20.0、50.0、100.0 μg/kg;由于油脂中普遍含有维生素E,故维生素E回收率采用试剂空白加标进行测试。
按1.2方法进行预处理,表3为各脂溶性维生素回收率结果。结果表明:视黄醇的回收率在97.0%~102.3%;视黄醇醋酸酯的回收率在98.1%~102.3%;维生素D2的回收率在96.3%~106.6%;维生素D3的回收率在96.7%~103.2%;维生素E的回收率在93.1%~99.1%。
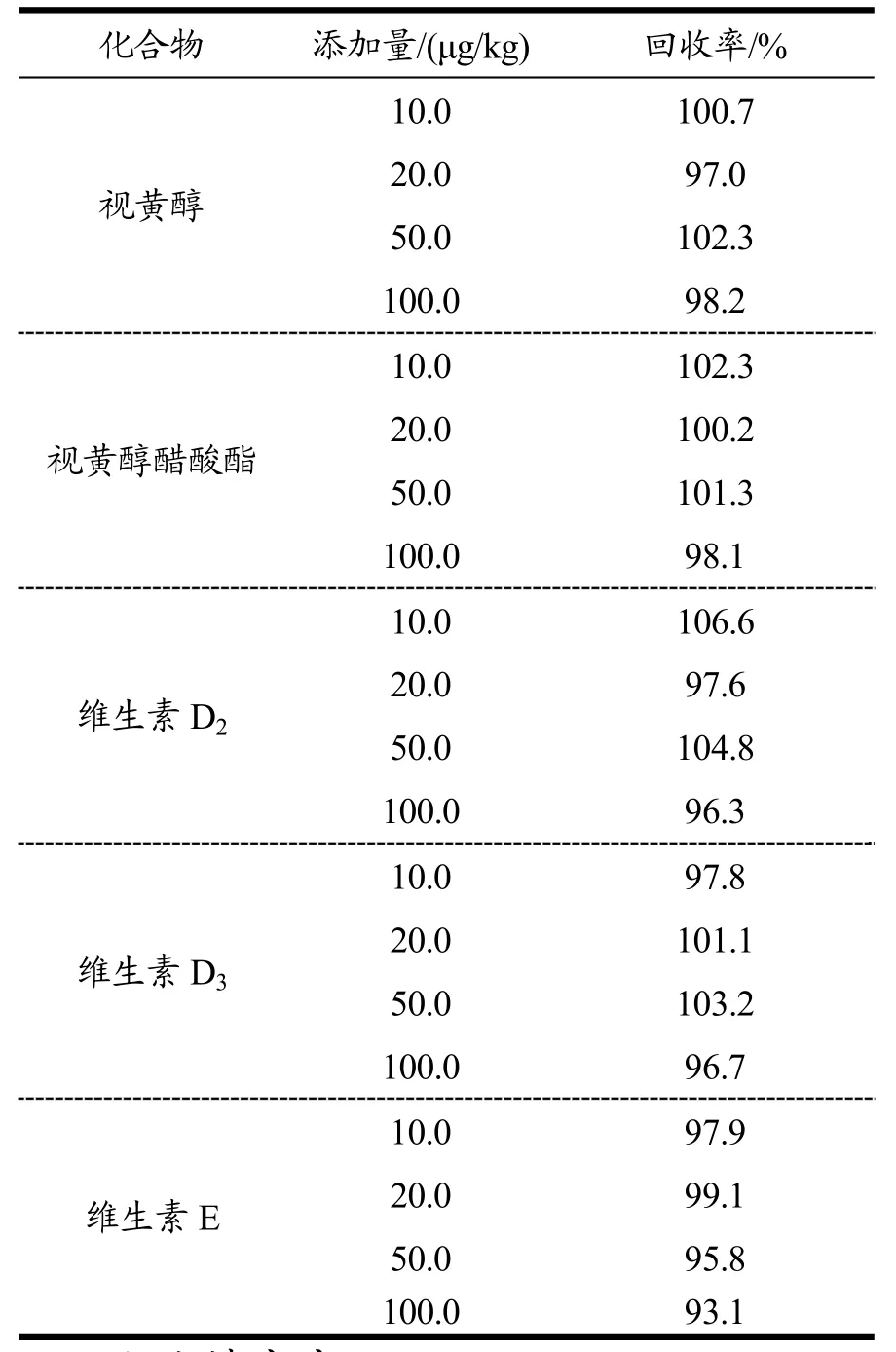
表3 各脂溶性维生素回收率结果Table 3 Results of recoveries of fat soluble vitamins
2.3.3 方法精密度
在不含有脂溶性维生素的大豆油基质中添加脂溶性维生素混合标准样品溶液,使大豆油中各脂溶性维生素含量为20.0 μg/kg,维生素E采用试剂空白加标进行测试。
按 1.2方法进行预处理,平行处理 6次,进行LC-MS/MS分析,测定结果见表4。结果表明:视黄醇、视黄醇醋酸酯、维生素D2、维生素D3、维生素E的相对标准偏差分别为:2.77%、1.09%、4.77%、2.13%、2.60%。
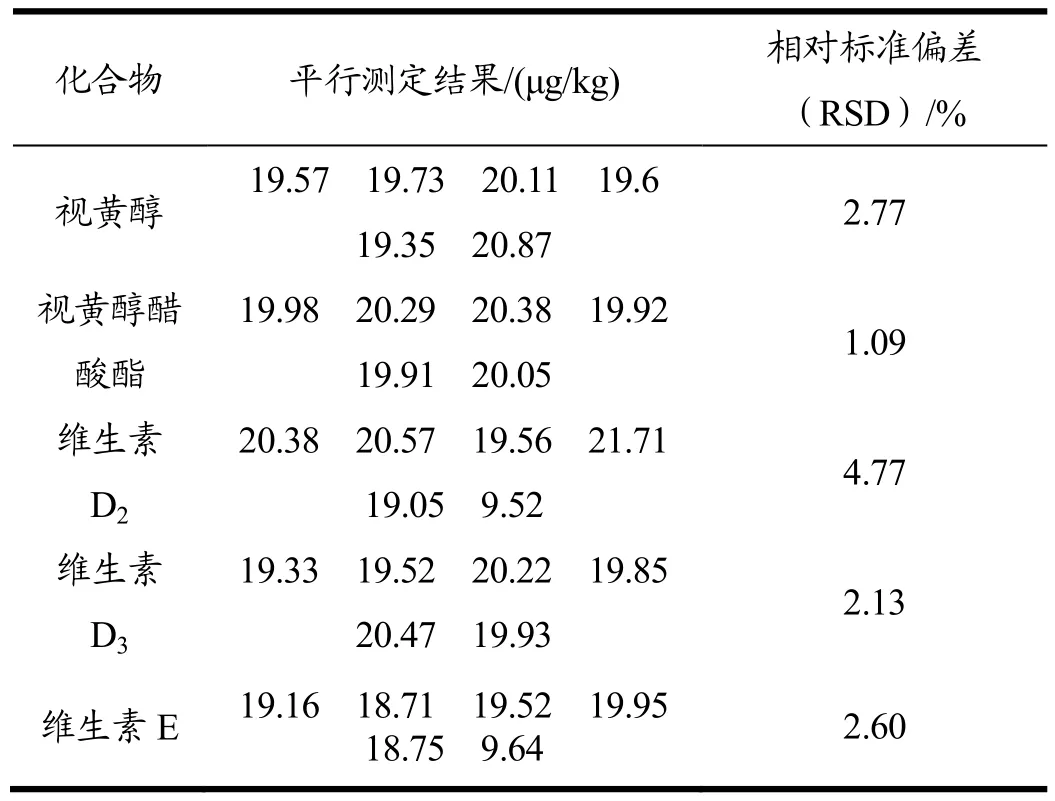
表4 脂溶性维生素精密度测试结果Table 4 Precision of fat soluble vitamins (n=6)
2.4 与皂化后液液萃取-液相色谱法的比较
2.4.1 检测对象比较
两种方法在检测对象上存在差异:皂化-液液萃取法在皂化处理过程中将维生素A、维生素E的酯水解为单体形式,统一了脂溶性维生素的检测形式,因此无需配备各种酯化标准样品。GPC为一种物理分离,在净化过程中不改变维生素的化学形式,在检测时需要了解维生素的添加形式,以选择合适的标准样品进行定性定量比较。
2.4.2 检测方法学指标比较
对凝胶渗透色谱-液相色谱串联质谱联用法(方法一)与皂化-液液萃取液相色谱法(方法二)测定油脂性食品中脂溶性维生素含量方法进行比较,方法学指标比较结果见表 5。结果表明:方法一与方法二在一定浓度范围内都有较好的线性关系,但方法一的检出限、回收率和精密度明显优于方法二。
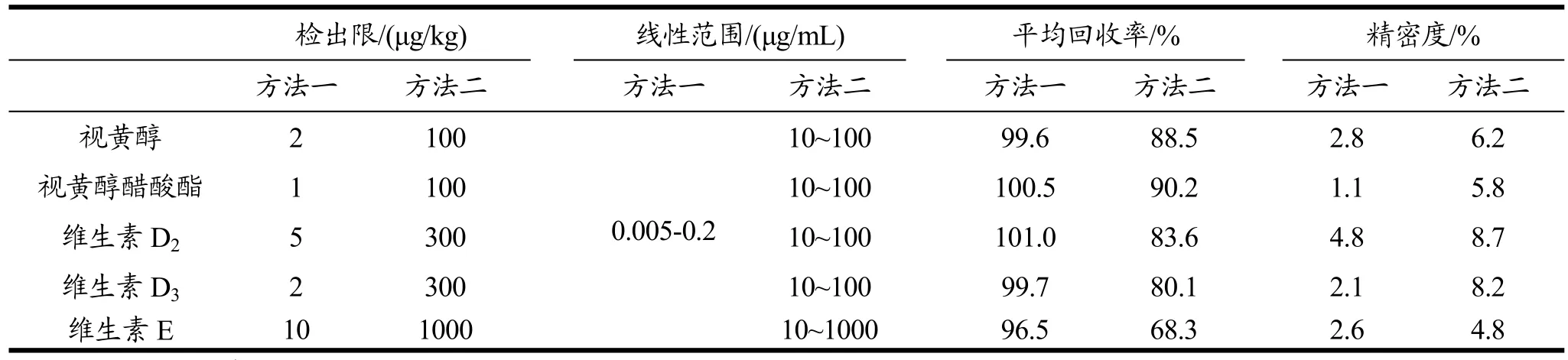
表5 方法学指标比较结果Table 5 Comparison of methodology index
2.4.3 检测效率比较
采用方法一前处理环节需要30 min,质谱分析时间为15 min;方法二前处理环节需要皂化45 min、冷却10 min、液液萃取及去除水分环节约需45 min、浓缩定容20 min、液相色谱分析时间为70 min。脂溶性维生素标准样品液相色谱图见图3。
从单个样品总分析时间比较方法一和方法二单个样品净分析时间分别为:45 min和170 min;从人力消耗上比较,方法一前处理环节可实现完全自动化、方法二除皂化过程外均需要人工操作。

图3 为脂溶性维生素标准样品液相色谱图Fig.3 Liquid chromatogram of fat soluble vitamins standard sample
3 结论
以PT785 Bio-BeadsS-X3(填料质量50 g)作为GPC分离色谱柱,环己烷-乙酸乙酯(5:5,V/V)溶液作为GPC流动相,流速为5 mL/min分离净化食用植物油、动物脂肪、油脂性保健食品中脂溶性维生素。以0.1%甲酸5 mmol/L乙酸铵水溶液和甲醇作为质谱流动相,结合ESI源,在正离子扫描模式条件下,采用LC-MS/MS同时测定维生素A、D、E含量结果满意,该法与皂化液液萃取-液相色谱法测定油脂性食品中维生素 A、D、E含量方法相比具有准确性高、灵敏度高、自动化程度高和分析时间短等多重优势。
