基于通路分析的重型痤疮全基因组关联研究
2018-10-12杨健康冯家祺张平曹光琼何黎吴文娟
杨健康 冯家祺 张平 曹光琼 何黎 吴文娟
671000云南,大理大学基础医学院(杨健康);昆明医科大学第一附属医院皮肤科(冯家祺、何黎、吴文娟),体检中心(曹光琼);云南曲靖市第一人民医院检验科(张平)
痤疮是一种常见的皮肤疾病,重型痤疮是痤疮中最严重的一类,它主要表现为广泛分布的炎症性皮损,经常导致患者的心理问题[1⁃2]。遗传因素在重型痤疮的发生中发挥重要作用[3]。全基因组关联研究(GWAS)是一种发现疾病易感基因的有效方法[4⁃5]。针对中国人群重型痤疮的GWAS研究曾发现2个新的易感基因[6]。但是,基因通常通过基因通路的形式在疾病的发生、发展中发挥作用[7]。因此,有必要进行基因通路分析,研究基因通路与疾病的关系,从而为全基因组关联研究提供有益补充。为了研究基因通路与重型痤疮的关系,我们进行重型痤疮全基因组通路分析,希望提高对重型痤疮发病机制的认识。
材料与方法
一、 数据
分析来源于重型痤疮全基因组关联研究的单核苷酸多态性(SNP)芯片数据[6]。采用美国Illumina公司HumanOmniZhongHua⁃8型芯片,于安徽医科大学皮肤病学教育部重点实验室行芯片扫描。数据集包括1 056例重型痤疮患者和1 056例健康对照的芯片数据,每个样本检测900 015个SNP位点。所有的重型痤疮患者都经皮肤科副主任或主任医师诊断为4型(Pillsbury分级系统):除颜面及胸背部泛发的粉刺、丘疹、脓疱外,皮损炎症重,并有囊肿、结节和瘢痕。本研究经昆明医科大学第一附属医院医学伦理委员会批准,患者及健康对照均签署知情同意书。标本由昆明医科大学第一附属医院、云南曲靖市第一人民医院、安徽医科大学第一附属医院等全国三十余家医院合作收集完成。
二、 方法
1.数据质控:对原始的SNP芯片数据进行严格的质量控制,以去除不合格的样本和SNP位点。首先对标本进行质控,排除检出率(call rates)<98%的样本,剩下合格的1 031份病例样本和1 031份健康对照样本。接着对SNP位点进行质控,①排除所有样本中检出率<98%的SNP位点;②排除所有样本中最小等位基因频率(MAF)<0.01的SNP位点;③排除健康对照样本中严重偏离哈迪-温伯格平衡(P< 1× 10⁃4)的SNP位点。经过质控,共筛出合格的SNP位点809 305个。
2.SNP位点分析:使用Plink v1.90软件[8]对SNP位点进行logistic回归分析。使用Q⁃Q图评估群体分层的大小及对结果的影响。使用Haploview v4.2软件做全基因组曼哈顿图。
3.基因分析:使用VEGAS软件计算[9],采用蒙特卡罗(Monte Carlo)法统计基因与重型痤疮的相关性,P值代表基因与重型痤疮相关的显著性水平。为了包括基因调控区的SNP位点,将基因的范围定义为编码序列(CDS)和20 kb的5′UTR及20 kb的3′UTR。接着将SNP定位到基因上。计算P值时,考虑了同一基因上所有SNP位点的影响,并且使用HapMap CHB和JPT的数据进行连锁不平衡校正。
4.通路分析:使用在线工具WebGestalt进行基因通路分析[10]。该基因通路为KEGG数据库定义的基因通路。为了减少通路大小对结果的影响,仅对包括10~200个基因的通路进行分析。采用超几何分布检验计算每条通路的P值,使用错误发现率(FDR)对得到的结果进行多重检验校正,校正后P<0.05视为通路与重型痤疮相关。
结 果
一、 SNP位点分析结果
40 920个SNP位点的P值小于0.05,见全基因组曼哈顿图(图1)。从图2可以看出所有SNP位点对应的P值整体上没有偏离零假设分布,提示病例对照样本匹配良好,人群分层因素对结果影响较小,与重型痤疮显著相关的SNP位点应该是真实的相关信号。
二、 基因分析结果
所有基因中,919个对应的P值小于0.05,其中12个P值小于0.001,见表1。
三、 通路分析结果
发现5条显著的基因通路(P<0.05),见表2。根据KEGG通路分类,可以将这5条通路分为代谢(1条)、内分泌系统(1条)、癌症(1条)、免疫系统(2条)。

图1 全基因组关联分析曼哈顿图 不同的颜色分别代表23对染色体;纵坐标为每个单核苷酸多态性(SNP)位点的-log10(P)值,该值越大代表该位点的等位基因频率在病例组与对照组间的差异越显著
讨 论
查询PubMed数据库和CNKI数据库,截至2018年6月,未检索到重型痤疮全基因组通路研究的相关文献,本研究可能是国际上第一个重型痤疮的全基因组通路研究,将有利于加深我们对于重型痤疮发病机制的认识。本研究发现的通路中有2个与免疫反应和炎症相关,分别是丙型肝炎和高亲和力IgE受体信号通路。这和目前普遍认为重型痤疮是炎症性疾病的认识一致[11⁃12]。研究认为,炎症反应会刺激毛囊皮脂腺导管角化过度,从而影响皮脂的正常排出[13]。
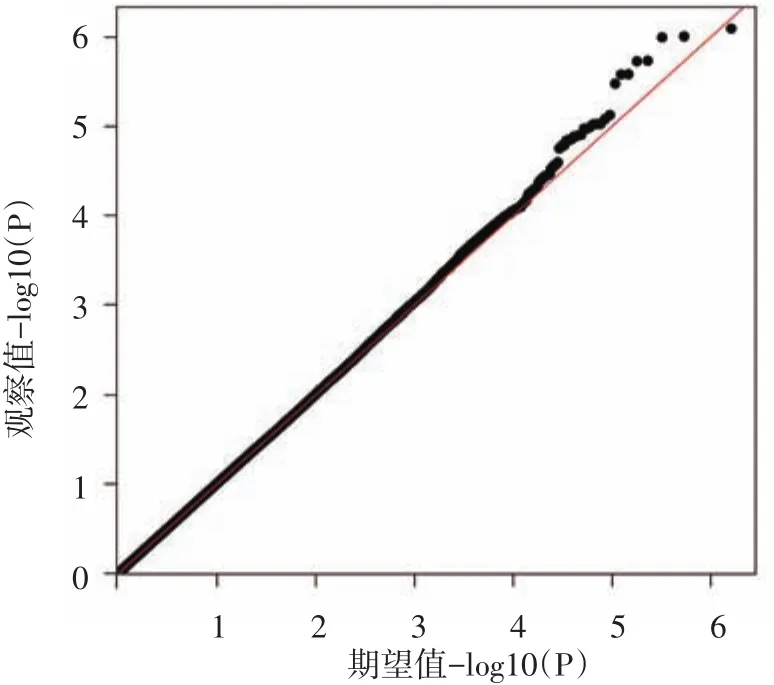
图2 全基因组关联分析Q-Q图 横坐标为809 305个单核苷酸多态性(SNP)位点-log10(P)值的期望值,纵坐标为-log10(P)值的观察值,图中观察值与期望值没有偏离零假设分布,提示病例对照样本匹配良好,人群分层因素对结果影响较小
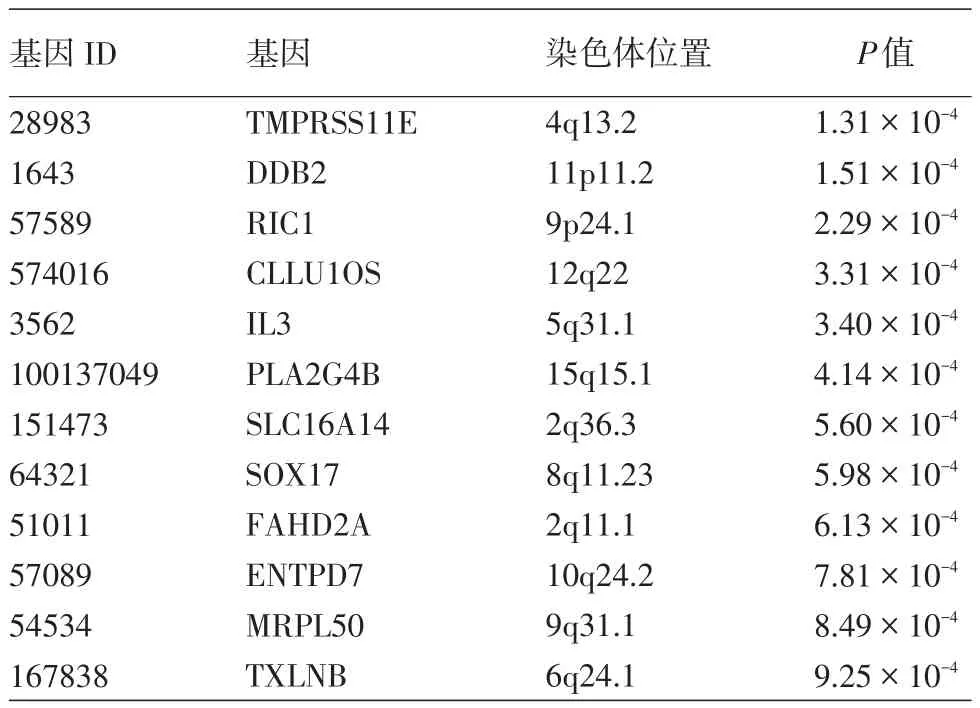
表1 基因分析发现的12个与重型痤疮关系最密切的基因

表2 通路分析方法发现的与重型痤疮相关的5条基因通路
本研究发现一些新的基因通路可能与重型痤疮相关,提示除了炎症、免疫相关通路以外,其他通路可能也参与重型痤疮的发生和发展。比如酪氨酸代谢通路,酪氨酸对于神经递质多巴胺的合成也发挥重要作用[14]。多巴胺是可以使人产生快乐的神经递质[15]。一项针对新加坡青少年进行的研究发现,痤疮的严重程度与精神压力呈正相关[16]。因此,酪氨酸代谢可能对于重型痤疮患者的精神压力也有重要的影响[17]。催乳素信号通路和肾细胞癌通路与痤疮的关系未见相关文献报道,需要进一步的功能研究来明确。
本次基因分析发现12个基因的P值小于0.001,其中包括重型痤疮全基因组关联研究发现的DDB2基因。DDB2基因编码的蛋白是一个新发现的与雄激素受体有相互作用的蛋白,可能通过泛素化使雄激素受体蛋白被蛋白酶体降解[18]。其他基因如CLLU1OS、IL3与炎症相关,IL3在激活与调节免疫细胞和介导T、B细胞增殖与分化及痤疮的炎症反应中起重要作用[19]。
本研究结果只是针对中国人群重型痤疮GWAS研究的全基因组通路分析,部分结果也可能存在假阳性的问题,有待新的重型痤疮GWAS研究对这些结果进行验证。我们发现一些可能与重型痤疮发病相关的基因通路,有望为后续的研究提供一些思路。
