结直肠腺瘤与息肉患者肠道菌群变化的病例对照研究
2018-10-10张永镇于恩达闫飞虎蒋露芳王和兴刘健翔李兆申蔡全才姜庆五
于 鑫 张永镇 于恩达 王 娜 帅 群 闫飞虎 蒋露芳王和兴 刘健翔 陈 跃 李兆申 蔡全才△ 姜庆五
(1复旦大学公共卫生学院流行病学教研室-教育部公共卫生安全重点实验室,5营养与食品卫生教研室-教育部公共卫生安全重点实验室 上海 200032; 2海军军医大学附属长海医院消化内科,6普外科 上海 200433; 3中国人民解放军92914部队医院消化内科 临高 571833;4渥太华大学医学系流行病与公共卫生学院 渥太华 K1G 5Z3)
结直肠癌是常见的消化道肿瘤之一,2013年中国结直肠癌发病人数居恶性肿瘤第4位,新发病例数约34.8万人,死亡人数约16.5万人[1]。随着人口老龄化、社会经济发展及生活方式改变等一系列原因,我国的结直肠癌发病率和死亡率呈逐年上升的趋势[2]。
结直肠癌的发生、发展与遗传和环境因素有关。85%~95%的病例以各种环境因素为病因,包括西方模式的饮食习惯、吸烟、饮酒、肥胖以及糖尿病等[3]。近年来,肠道菌群改变以及促肿瘤微环境的形成在结直肠癌发生、发展中的作用日益受到关注[4-5]。
主流观点认为,大部分结直肠癌的发展经历了正常上皮细胞—腺瘤—进展期腺瘤—肿瘤的一系列过程[4]。已有动物实验证据表明特定细菌(如具核梭杆菌)与结直肠肿瘤的形成密切相关[6],然而关于良性的增生性息肉与腺瘤和作为癌前病变的进展期腺瘤的菌群变化特征研究较少,样本量也较小。本研究基于单中心病例对照研究,分析结直肠癌早期病变(癌前病变与良性病变)患者的肠道菌群变化特征,探索在结直肠癌早期病变中可能发挥重要作用的细菌,为肠道菌群变化与结直肠癌早期阶段的关系提供线索。
资 料 和 方 法
研究对象本研究为病例对照研究,研究对象来自2014—2015年海军军医大学附属长海医院门诊的肠镜受检者。共纳入研究对象280例,包括88例进展期腺瘤患者、32例单纯腺瘤患者、30例增生性息肉患者以及130位无结直肠疾病的受检者。将进展期腺瘤患者纳入癌前病变组;将单纯腺瘤患者与增生性息肉患者纳入良性病变组;将无结直肠疾病的受检者纳入对照组。
病例组研究对象纳入标准:(1)患者在长海医院初次被诊断为相应疾病;(2)年龄≥40岁;(3)汉族;(4)检查前无肠道肿瘤、息肉、炎性肠病,无结直肠肿瘤家族史,无其他器官肿瘤病史和肠道相关手术史;(5)过去6个月未使用过抗生素;(6)能同时获得治疗前粪便标本并填写调查问卷表;(7)对参加本课题研究知情同意。对照组经结肠镜检查无结直肠相关疾病,其余纳入标准与病例组一致。本项研究通过海军军医大学伦理委员会审核(编号:CHEC2015-082)。
样本收集与处理所有研究对象均完成一份调查问卷,包括年龄、性别、体重指数(body mass index,BMI)、生活习惯(包括吸烟、饮酒)、疾病既往史等。BMI≥24 kg/m2为超重,BMI≥28 kg/m2为肥胖[7]。位于盲肠、升结肠、肝曲、横结肠、脾曲的病变为近端病变,而位于降结肠、乙状结肠与直肠为远端病变。进展期腺瘤指直径超过1cm的腺瘤、绒毛状腺瘤或伴有重度不典型增生的管状腺瘤。吸烟、饮酒情况按照从无或曾经有过进行分类。
所有研究对象的新鲜粪便样本(≥1 g)于结直肠镜检肠道准备之前取得。粪便样本细菌的DNA使用OMEGA-soil DNA试剂盒(美国Omega Bio-Tek公司)按照说明书进行提取。经过PCR扩增之后,对细菌的16S rDNA的V3~V4片段进行测序,使用通用引物338F 5’-ACTCCTACGGGAGG-CAGCAG-3’与806R 5’-GGACTACHVGGGTW-TCTAAT-3’。测序由上海美吉生物医药科技有限公司在Illumina MiSeq平台完成。
生物信息学分析与统计分析采用 Trimmomatic软件筛除序列尾部质量值20以下的碱基,过滤50 bp窗口内平均质量低于20的窗口以及尾部,筛除单条序列,在此基础上以97%相似度聚类得到操作分类单元(operational taxonomic units,OTU),挑选出每个OTU的代表序列并利用SINTAX算法[8]与RDP数据库对比得到每个OTU的群落名称。参数设置:对门、纲、目、科、属级别的置信阈值为0.8,对种级别的置信阈值为0.5。
为比较不同组的研究对象肠道菌群物种多样性,基于OTU表格计算α多样性(ace,shannon,simpson,PD whole tree)与β多样性(非加权和加权Unifrac距离),并对3组研究对象的α多样性进行方差分析。为衡量总体的菌群结构差异,基于β多样性进行PERMANOVA分析,同时控制潜在的混杂因素,包括年龄、性别、BMI、吸烟及饮酒情况。
为比较3组研究对象在属与种两个分类级别上的差别,使用怀特非参数检验[9](White’s non-parametrict-test)挑选出差异细菌并进行可视化,多重比较校正采用Benjamini-Hochberg方法。生物信息分析利用QIIME 1.9.1平台完成,统计分析使用R软件v 3.3.2完成,不同组间细菌丰富度差异的检验应用STAMP v 2.1.3软件。
结 果
研究对象的一般情况研究对象的年龄为40~84岁,癌前病变组、良性病变组和对照组在性别、年龄、BMI和病变位置方面的差异均无统计学意义,癌前病变组与良性病变组相比,病变部位更多位于远端(χ2=4.84,P=0.028)。
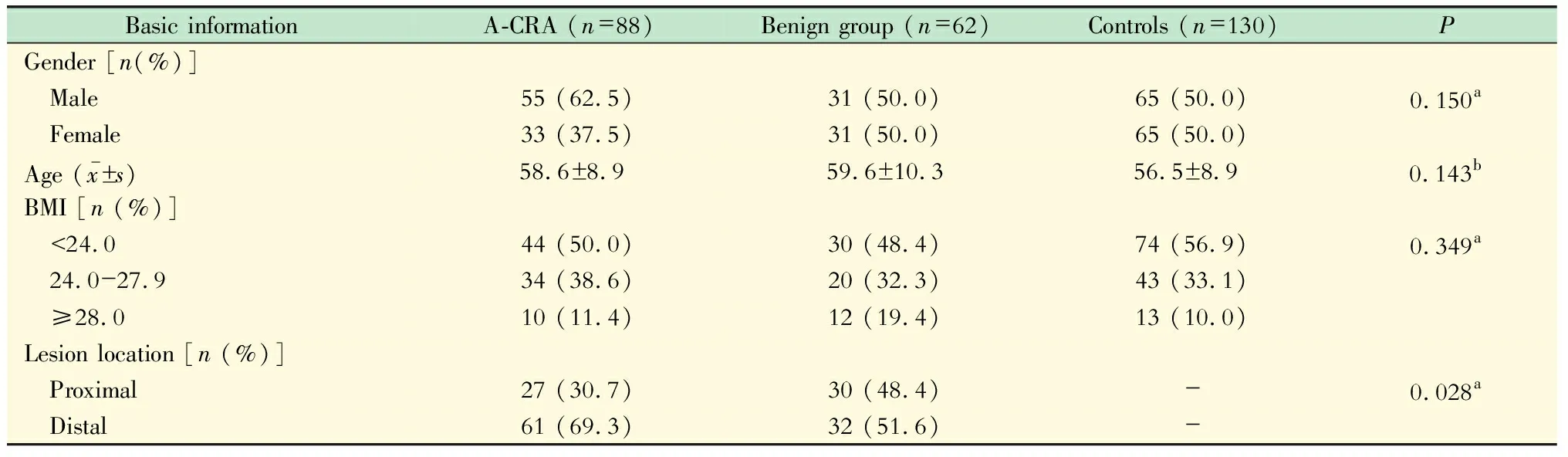
表1 结直肠腺瘤与息肉患者及对照组的一般情况Tab 1 Distribution of basic information among patients with and without colorectal adenoma and polyps
aPvalues were calculated byχ2test;bPvalue was calculated by ANOVA;A-CRA:Advanced colorectal adenoma;Polyps:Colorectal adenoma and hyperplastic polyps.
肠道菌群总体描述经过质量控制与清洗,最终在280个样本中得到19 714 595条高质量序列,1 512个OTU。根据物种分类可以分为12个门,19个纲,29个目,57个科,119个属,238个种(纳入条件:门平均相对丰富度> 0.01%;纲至种:相对丰富度>0.001%)。
三组样本的α多样性无统计学差异(ace:F=2.293,P=0.103;shannon:F=0.079,P=0.924;simpson:F=0.181,P=0.835;PD whole tree:F=1.540,P=0.216;方差分析)。在控制了年龄、性别、BMI以及吸烟饮酒情况之后,基于β多样性距离进行PERMANOVA分析发现,不同组的肠道菌群结构有统计学差异(非加权Unifrac 距离:F=1.834,P=0.005;加权 Unifrac 距离:F=0.725,P=0.626)。
癌前病变组、良性病变组与对照组在“属”分类级别的比较通过对119个菌属进行非参数检验,结果显示共有7个菌属在早期病变患者体内有显著变化。与对照组相比,癌前病变组患者体内的卟啉单胞菌属(Porphyromonas)(P<0.001)与Alloprevotella(P<0.001)明显上升,而双歧杆菌属(Bifidobacterium)显著下降(P=0.018);良性病变组患者Akkermansia(P=0.007)、嗜血杆菌属(Haemophilus)(P<0.001)和乳酸杆菌属(Lactobacillus)(P<0.049)显著增多,而克雷伯氏菌属(Klebsiella)(P<0.001)和双歧杆菌属(P<0.001)显著减少(图1)。
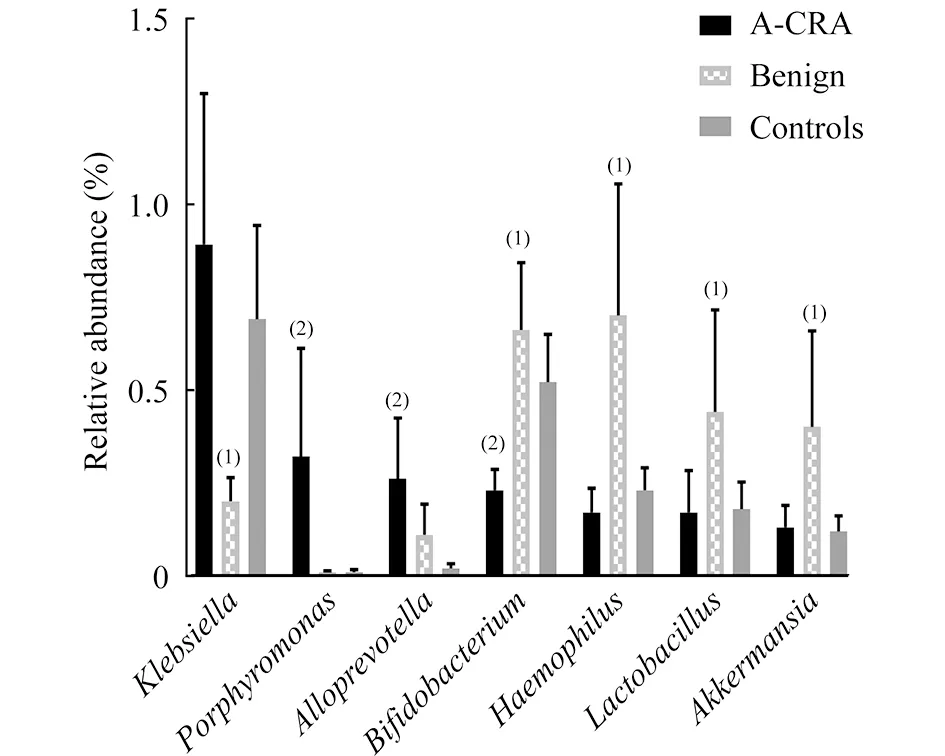
(1)polypsvs.control,(2)A-CRAvs.control,P<0.05.
图1结直肠腺瘤与息肉患者与对照组相比在“属”级别细菌变化情况
Fig1Featuredmicrobesinthecomparisonbetweencontrolsandpatientswithcolorectaladenomaandpolypsatthegenuslevel
癌前病变组、良性病变组与对照组在“种”分类级别的比较对238个菌种进行非参数检验,发现与对照组相比,癌前病变组患者粪便中有11个菌种存在显著差别(图2),其中Clostridiumbolteae(P=0.014)、Alloprevotellarava(P<0.001)、Enterobactersoli(P<0.001)、牙龈卟啉单胞菌(Porphyromonasgingivalis)(P<0.001)共4种细菌显著上升,而丁酸梭菌(Clostridiumbutyricum)(P=0.001)、Veillonelladispar(P<0.001)、Veillonellarogosae(P=0.005)、Fusobacteriummortiferum(P<0.001)、Bacteroidescoprophilus(P=0.026)、Clostridiumcolinum(P=0.030)与Streptococcusdentisani(P=0.028)共7种细菌中显著下降。
与对照组相比,良性病变组患者粪便内发现副流感嗜血杆菌(Haemophilusparainfluenzae)(P<0.001)、Prevotellastercorea(P=0.011)、Clostridiumbolteae(P=0.006)、Enterobactersoli(P<0.001)、Akkermansiamuciniphila(P=0.006)、Veillonellaatypica(P<0.001)、Veillonellaratti(P=0.007)共7种细菌显著上升,而Veillonelladispar(P<0.001)、Megasphaeraindica(P=0.014)、Clostridiumcolinum(P=0.027)和Clostridiumbutyricum(P=0.007)共4种细菌显著下降(图2)。
讨 论
本研究通过以医院资料为基础的病例对照研究,对首次诊断的进展期腺瘤、单纯腺瘤和增生性息肉患者肠道菌群构成变化进行了分析。
近年来,关于肠道菌群的变化与结直肠癌的发生发展关系的重要研究不断增多[4-5,10]。本研究比较进展期腺瘤患者与对照组的粪便菌群发现,卟啉单胞菌属以及其菌种牙龈卟啉单胞菌、Alloprevotella以及其菌种Alloprevotellarava在进展期腺瘤患者粪便中明显上升。牙龈卟啉单胞菌是一种能引起牙周疾病的厌氧致病菌,与口腔癌的发生密切相关。流行病学研究发现[11],牙周病以及牙齿牙列缺失可能增加结直肠癌的发生风险。有研究指出[12],卟啉单胞菌属、消化链球菌(Peptostreptococcus)等引起牙周疾病的致病菌在结直肠癌患者肠道菌群中有显著上升,猜测牙周致病菌同时是结直肠癌潜在致病菌,牙龈卟啉单胞菌等致病菌对结直肠癌的致病模式可能与牙周病类似。提示牙周病致病菌对结直肠癌可能有重要作用。关于Alloprevotellarava的研究较少,有文献指出其与白塞病可能有关[13]。
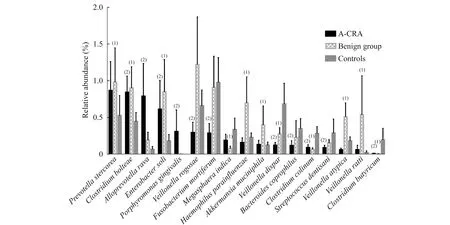
(1)Polypsvs. control,(2)A-CRAvs. control,P<0.05.
图2结直肠腺瘤与息肉患者与对照组相比在“种”级别细菌变化情况
Fig2Featuredmicrobesinthecomparisonbetweencontrolsandpatientswithcolorectaladenomaandpolypsatthespecieslevel
双歧杆菌属是人体肠道内重要的常见共生细菌。该菌属下的一些菌种被认为是能对机体产生有利影响的益生菌[14]。既往研究中指出,在结直肠癌患者粪便中以及肠道黏膜附着的双歧杆菌属丰富度明显下降,提示其可能在结直肠癌进展过程中发挥预防与保护的作用[15-16]。双歧杆菌能发挥抗过敏效应、减少有害细菌等多种生理作用[17]。在本研究中,进展期腺瘤患者体内双歧杆菌属较对照组显著降低,与既往研究结论一致。
丁酸梭菌以及Streptococcusdentisani的变化同样值得关注。本研究发现,这两种细菌在结直肠癌早期病变患者体内显著下降,且随着病情发展细菌丰富度降低。丁酸梭菌是一种常见于人体肠道的严格厌氧菌细菌。有研究报道,小鼠口服丁酸梭菌能够调节消化道菌群,提升益生菌丰富度,并能通过引导F4/80+CD11b+肠道巨噬细胞产生IL-10从而抑制小鼠急性结肠炎[18-19]。实验研究显示,丁酸梭菌能起到减少肠道肿瘤发生的作用[20]。Strepto-coccusdentisani是一种存在于健康人牙菌斑内的正常细菌,研究显示其不产生毒素并且能够通过产生细菌素抑制主要口腔致病菌的生成以及缓冲过酸性pH条件,被认为是一种益生菌[21]。除此之外,属于人体肠道常见共生菌的韦荣球菌属[22]的菌种(如Veillonelladispar)也被发现在进展期腺瘤患者粪便中减少。
本研究观察到进展期腺瘤和良性病变患者的α多样性与对照组相比差异无统计学意义,且加权Unifrac距离并未显示菌群存在结构变化。造成该结果的原因可能是,粪便菌群与病变黏膜细菌相比,受到更多因素的影响[10]。
本研究结果显示,大部分在患者体内有显著差异的细菌并非随疾病恶性程度丰富度增加或减少。在良性病变患者体内发现克雷伯氏菌属、乳酸杆菌属、双歧杆菌属、Akkermansia等细菌显著升高的现象,猜测可能是由于良性病变导致了某些益生菌代偿性地增加。由于目前对良性病变患者肠道细菌变化规律和特定细菌生理特性的研究较少,尚无法判断出现此情况的具体原因。
综上所述,本研究发现进展期腺瘤、良性病变(单纯性腺瘤与增生性息肉)患者体内肠道菌群与对照组相比在整体结构和多种细菌上有显著的变化,进展期腺瘤患者体内牙周病致病菌丰富度上升、益生菌丰富度下降,良性病变患者体内多种益生菌丰富度发生变化。上述变化可能与结直肠癌早期病变的发生发展有关。本研究对结直肠癌早期病变的肠道菌群变化规律做了初步探索,为结直肠癌病变的早期阶段与肠道菌群变化的关系提供了线索。
