Ce掺杂改性Ni-Al-Ox催化剂CO-NO反应性能
2018-09-25常化振李俊华
郭 磊,张 涛,常化振*,李俊华
Ce掺杂改性Ni-Al-O催化剂CO-NO反应性能
郭 磊1,张 涛1,常化振1*,李俊华2
(1.中国人民大学环境学院,北京 100872;2.清华大学环境学院,北京 100084)
通过尿素法制备了一系列Ce掺杂改性的Ni-Al-O复合氧化物催化剂,并研究了催化剂的CO-NO反应性能.活性测试结果表明,Ni-Al-Ce-O的催化活性明显优于Ni-Al-O,且活性随着Ce含量的增加而提高.当Ce的掺杂量为20%时,250℃条件下,NO转化率高达95%以上,同时催化剂具有良好的抗H2O性能. XRD和Raman分析表明, Ce掺杂Ni-Al-O催化剂促进了氧空位产生.而XPS结果显示,氧空位随着Ce含量的增加而增加.H2-TPR结果表明,Ce的掺杂使催化剂的氧化还原性能增强.结合NO-TPD和FTIR的表征结果,进一步发现Ce的加入使表面氧难以将NO氧化成硝酸盐,有利于NO在Ce的氧空位上分解为N2或N2O,促进CO-NO反应的进行.
Ni-Al-Ce-O;CO-NO反应;氧空位;Ce掺杂
近年来,大气污染物排放标准日趋严格,对机动车尾气净化要求越来越高,尾气中氮氧化物(NO)等的高效净化成为一个亟待解决的问题[1-4].CO-NO反应能同时去除尾气中的CO和NO,是消除尾气污染物的理想方法.目前,在CO-NO反应中应用较成熟的催化剂是贵金属催化剂,但存在价格昂贵,高温不稳定等缺点.因此,用更为经济的过渡/稀土金属氧化物取代贵金属在机动车尾气净化领域一直是人们关注的方向.
类水滑石为前驱体制备的复合氧化物具有较大的比表面积和良好的氧化还原性能,近年来, 逐渐成为最具有开发前景的消除NO的催化剂之一.目前类水滑石结构催化剂在脱硝领域的应用主要体现在NH3-SCR,NO储存以及CO-NO反应中.在NH3-SCR的研究中发现,过渡金属Cu,Co掺入水滑石前驱体或碱土金属Mg和两性金属元素Al作为水滑石前驱体可以显著提高制得的催化剂的NH3- SCR催化活性.[5-6]在NO吸附存储的应用中, Co,Mg, Al元素也是很好的水滑石材料. Basile F等用水滑石焙烧得到的MgAl复合氧化物为载体,采用浸渍法得到了系列Pt和Pt-Cu负载的NO存储还原催化剂,该系列催化剂低温时的NO存储活性大大提高,并且具有较强的抗SO2能力[7].Yu等[8]发现CoMgAl水滑石具有良好的NO吸附储存性能.此外,研究发现碱金属K可以显著提高CoMgAl水滑石的NO储存性能[9].类水滑石在CO-NO反应中也展现出了良好的催化性能.陈英红等将CoNiAl类水滑石复合氧化物催化剂用于CO-NO反应,发现当Co:Ni: Al=5:1:1(物质的量之比)时,催化剂在低温下有一定的反应活性,200℃的NO转化率达到95%,但温度继续升高,活性明显下降[10].Dai等[11]发现向CoMgAl类水滑石复合氧化物催化剂中添加Ce能明显提高催化剂的氧化还原能力,促进其NO脱除性能.Palomares等[12]利用CoMgAl类水滑石复合氧化物催化剂处理催化裂化流化床单元条件下所产生的NO时发现,CeO2的加入能明显提高催化剂的抗SO2能力.此外,已有研究报道,将Ce加入NiO中可以产生新的氧化还原位点,通过Ni和Ce之间的相互作用提升其CO-NO反应性能[13-14].总体而言,Co,Mg,Al是很好的水滑石构成元素,表现出在NH3-SCR、NO储存以及CO-NO反应中很好的性能.而Ce元素在CO-NO反应中可能具有很好的应用前景.
由以上可知,类水滑石催化剂在包括CO-NO反应的催化净化NO技术中有广泛运用,但也存在抗SO2性差,温度窗口较窄等缺点,而CeO2具有优异的储氧性能和氧化还原性,作为助剂可以有效提高催化剂的催化活性,更是能与Ni产生相互作用.因此,本文在以上研究的基础上,考察了Ce掺杂改性的Ni-Al-O复合氧化物催化剂对CO-NO反应的催化性能,并利用XRD、TPR、Raman等表征手段对Ce掺杂提升Ni-Al-O的CO-NO催化性能的原因进行了探究.以期为Ce在水滑石催化CO-NO反应中的作用机理的研究做出贡献.
1 实验部分
1.1 催化剂制备
按照Ni2++Al3++Ce3+= 1mol/L, (Ni2++Ce3+)/Al3+= 3配制镍盐和铝盐混合溶液A,尿素溶液B(尿素/(Ni2++Ce3++Al3+)=10).在室温和剧烈搅拌条件下,A和B同时滴加到盛有一定去离子水的三颈烧瓶中.滴加完转入烧杯中,放入95℃油浴中继续搅拌24h.冷至室温,5000r/min离心分离并用去离子水洗涤沉淀至混合液呈中性,80℃过夜烘干得前驱体.在500℃下煅烧4h得Ni-Al-Ce-O复合氧化物催化剂,以NiAlCeO表示,其中=Ce3+/(Ni2++Ce3+). Ni-Al-O催化剂的制备方法与此类似,制得的催化剂以NiAlO表示.
1.2 催化剂表征
XRD分析:本实验使用X射线衍射仪测定催化剂的晶相构成,仪器型号为D8Advance.其中管电压为40KV,Cu靶,扫描角度为10°~90°,8°/min连续扫描.依据谢乐公式(式1),计算催化剂晶粒尺寸.
式中:=0.89;为入射X射线波长,nm;为半峰宽,°;为衍射角,°.
H2-TPR:本实验使用AutoChem 2920进行测定.首先称取样品50mg,在N2氛围300℃下预处理1h.然后在10%H2中以10℃/min的速率从室温升到1000℃. Raman光谱测定:本研究中采用英国的雷尼绍(inVia)仪器进行Raman分析.
XPS分析:本研究中XPS分析是由PHI Quantera SXMTM(ULVAC-PHI Inc)仪器完成.采用Al/Mg双阳极靶,结果中各个元素的结合能需要校正(284.8eV C1峰校正).
NO-TPD:称取催化剂100mg,然后在300℃,200mL/min N2条件下进行预处理.之后将温度降至室温,通入500×10-6NO/N2吸附1h.然后用N2吹扫1h,最后以10℃/min从100℃升到500℃,记录脱附出NO浓度.
In situ DRIFTS表征:在NICOLET6700型傅立叶红外光谱仪上进行实验.将研细的样品填满样品池,通冷却水后,先在N2气氛下400℃预处理1h.然后在N2吹扫下采集背景图谱.切换至吸附气体100℃下吸附至饱和,N2吹扫至稳定,采集不同温度下的DRIFTS图谱.
1.3 CO-NO活性评价
活性评价在实验室自行设计的石英固定反应床上测试.利用瑞士Eco Physics公司的氮氧化物分析仪(型号:CLD 822Mh)检测气体浓度.反应条件如下:0.15g样品,500×10-6NO, 2000×10-6CO, 0或5%的H2O, N2平衡气,总气体流速为200mL/min.空速(GHSV)为90000h-1.NO转化率计算公式如下:

式中:[NO]in和[NO]out分别代表稳定状态下NO的进口和出口浓度.
2 结果与讨论
2.1 CO-NO的反应性能
图1是不同Ce比例掺杂的NiAlCemO催化剂的CO-NO反应活性曲线.结果表明,NiAlO的活性较低,掺杂5% Ce后,催化剂活性明显提高.Ce掺杂量提高后,催化剂的低温活性进一步提高. NiAlCe30O催化剂的活性最佳,250℃时NO转化率达到了95%.Ce的掺杂量再提高至40%后,反应活性开始下降.可以看出,Ce的掺杂能够提高催化剂的CO-NO反应活性.

图1 不同催化剂的CO-NO反应活性
反应条件为500×10-6NO,2000×10-6CO,N2平衡, GHSV = 9.0×104h−1(STP)
2.2 抗水性能探究
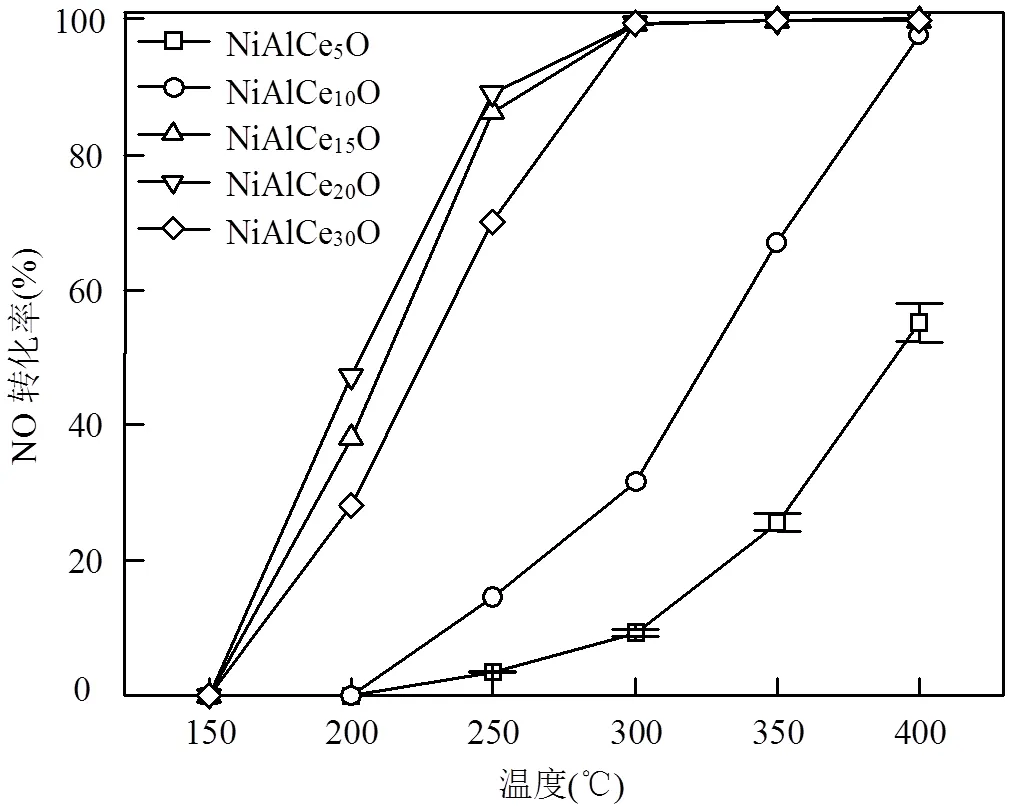
图2 NiAlCemO催化剂在CO-NO反应中的抗水性能
反应条件为0.2g 样品,500×10-6NO, 2000 ×10-6CO,5%H2O,N2平衡,GHSV = 9.0×104h−1(STP)
图2显示了制备的不同催化剂在H2O存在的情况下CO-NO反应活性的变化.对于NiAlCe5O和NiAlCe10O,活性受水的影响很大,而对于Ce含量较高的样品,仅低温区间有所影响,以NiAlCe20O为例,200℃时活性由80.5%下降至47.3%,但250℃以后便几乎不再受水的影响.总的来说,H2O在一定程度上会较大地抑制NiAlCeO的CO-NO氧化活性,但在一定范围内提高Ce的掺杂量确实能提高抗水性能.
2.3 NiCeO固溶体和氧空位的形成
2.3.1 XRD图3是不同催化剂的XRD图谱 NiAlO样品上的峰均归属于NiO的特征峰,没有Al的特征峰出现,说明Al在该氧化物中具有良好的分散度.掺杂Ce后出现CeO2的特征峰,且随着Ce的含量增加,CeO2的特征峰增强,NiO的特征峰减弱.与纯CeO2相比,NiAlCeO中的CeO2的特征峰都出现轻微偏移,这主要是由于Ce4+和Ni2+的离子半径差异较大,Ni物种进入CeO2后引起了后者的晶格畸变[15].这也说明Ni2+进入CeO2晶格形成了固溶体.因此,掺杂的Ce可能以两种物相存在:一部分与Ni2+相互作用形成固溶体,一部分以CeO2形式存在.
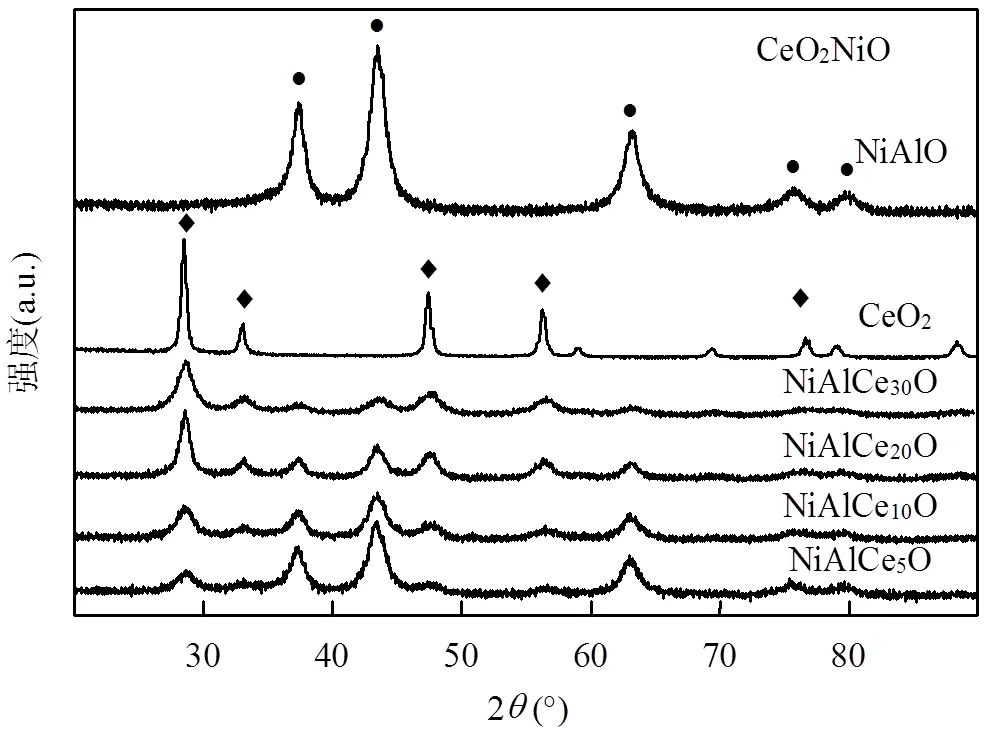
图3 NiAlO,NiAlCemO和CeO2催化剂的XRD图谱
2.3.2 Raman光谱 如图4所示, Ce掺杂量在10%及以上的样品在461cm-1出现了明显的尖峰,这是由[Ce-O8]的伸缩振动(又称F2g振动模式)引起[16].结合文献可知,CeO2原本位于465cm-1的特征峰向低波数移动,证明Ce-O键强度减弱,这极有可能是由于Ni2+进入Ce-O晶格形成的.为了维持电荷平衡,当Ni2+进入Ce-O晶格后,必然伴随氧空位的产生[17].位于227,634cm-1的拉曼特征峰则直接证明了氧空位和Ni-Ce-O固溶体的形成[18-19].位于500~ 600cm−1的对称拉曼特征峰归属于Ni-O键的振动[17].加入Ce后,该吸收峰向高波数移动,可能是CeO2上氧空位的峰(D)[19-21]与Ni-O键的振动峰重合.Raman光谱的结果不仅明确证明了Ni-Ce-O固溶体的产生,也给出了氧空位存在的证据.

图4 NiAlO和NiAlCemO催化剂的拉曼光谱
2.3.3 XPS 为了进一步探讨材料表面原子价态变化,接下来利用XPS分析对催化剂进行了表征.NiAlCeO催化剂样品的O 1s谱图如图5(a)所示.经分峰可知,结合能在531.4~531.8eV之间的峰可以归属于表面化学吸附氧,如O2-、O-,这些氧物种来自于氧空位或羟基类(记为O)[22];而结合能在529.8~530.1eV之间的峰可以归属于晶格氧(记为O)[23].通过积分计算可以得到O/(O+O)的比例(图中列出).显然,O/(O+O)的比例与Ce的掺杂量呈正相关.更多的化学吸附氧意味着催化剂表面存在更多可经CO还原产生的氧空位.同时,O 1s结合能的大小(具体见表1)跟Ce的掺杂量呈现负相关,这是由于Ce极强的电负性导致.根据以往的研究,同一体系内O 1s的结合能单调减小意味着O的碱性增强[24],不难推测Ce的添加增加了催化剂内O的碱性,使其在反应中更易吸附CO.这有利于NiAlCeO催化剂CO-NO反应活性的提高.
NiAlCeO催化剂 Ce 3d谱图如图5(b)所示.v和u分别代表3d5/2和3d3/2的自旋轨道耦合.v,v´´,v´´´,u,u´´和u´´´属于Ce4+的特征峰.v0,v´和u´属于Ce3+的特征峰[25]. Ce4+含量要高于Ce3+,说明在NiAlCeO催化剂中Ce4+和Ce3+共同存在,且Ce4+占据主导地位.这与已报道的结果一致[26].在该系列催化剂中,Ce3+/(Ce3++Ce4+)的比例与Ce的添加量成正相关(表1).相对较高的Ce3+比例意味着更多的氧空位[27].这可能是CO-NO活性随Ce含量增加的原因之一.
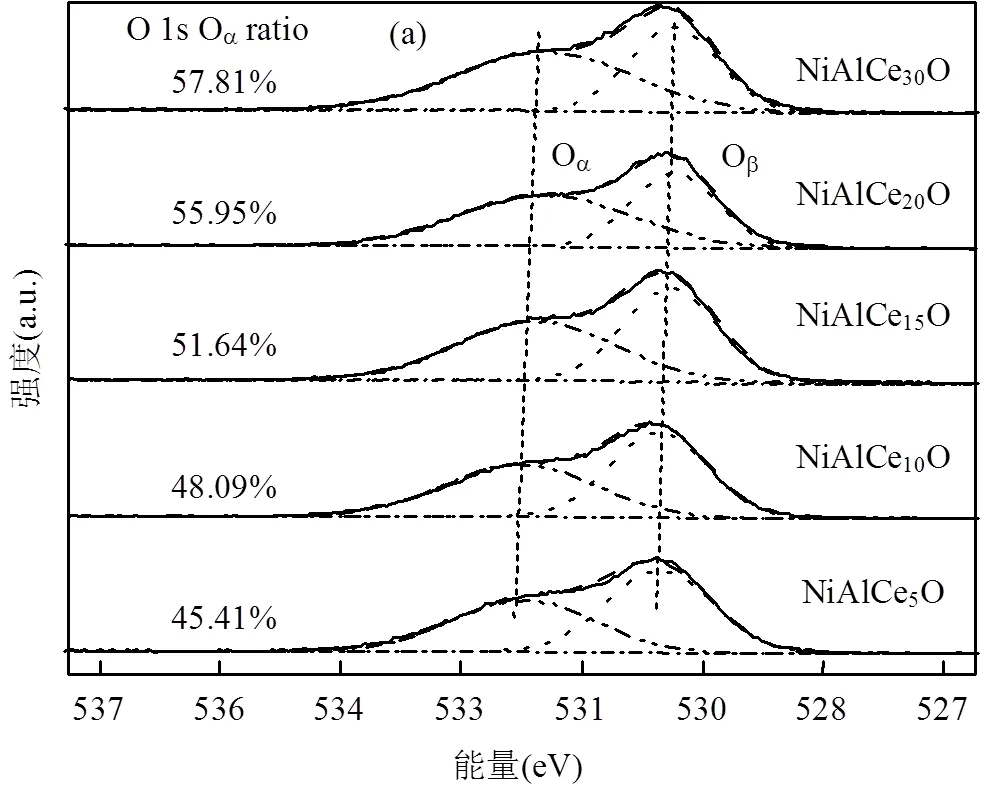

表1 NiAlCemO的耗氢量和XPS信息
注:-为未检验.
2.4 H2-TPR
如图6所示,所有催化剂在550℃附近均有一个明显的还原峰(α峰).对于NiAlO,α峰归属于高度分散的NiO被还原过程[28-29].而对于NiAlCeO,除了代表催化剂表面NiO被还原, α峰还表明进入CeO2晶格中的NiO的还原.显然,随着Ce掺杂量从10%上升到30%,还原峰逐渐向高温区移动.一般来说,高度分散的NiO可以在相对较低的温度下被还原,而进入固溶体的Ni物种则相对来说更难被还原[29].因此,随着Ce含量增加,越来越多的Ni进入CeO2晶格形成固溶体(XRD和Raman的结果有所证明),最终导致α峰向高温区移动.耗氢量的变化也说明了这一点(表1).此外,在该温度区间仅有一部分CeO2能被还原.对于NiAlCe5O,α峰出现的温度相对较高,可能是由于它拥有相对完整的水滑石结构,Ni2+在体系内的分散度更高,所以更难被还原[28].对于NiAlCe20O和NiAlCe30O,在300℃左右出现了一个新的还原峰,可能归属于CeO2上表面Ce4+的还原,这从侧面说明了Ni-Ce-O固溶体的形成[30].这也证明Ce的掺杂促进了两者的氧化还原性.而对于NiAlO在280℃出现的弱还原峰,可能属于体相NiO的还原[31].
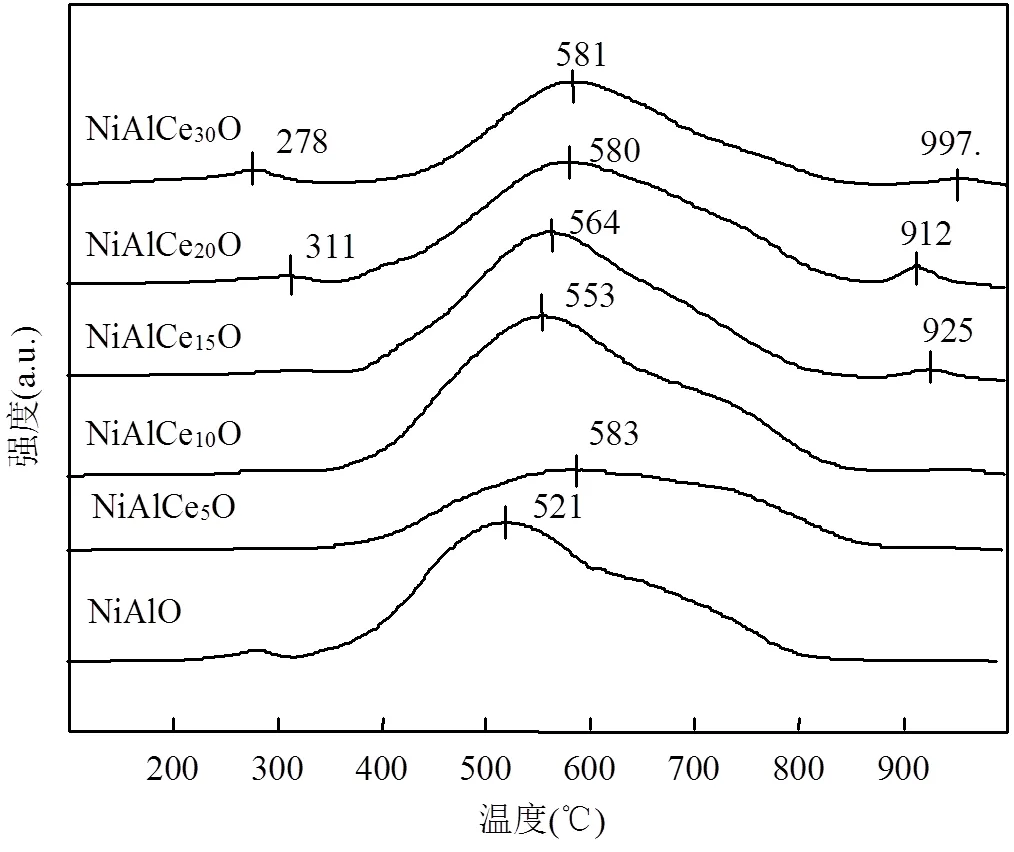
图6 NiAlO和NiAlCemO催化剂的H2-TPR图谱
2.5 NOx-TPD
为了评价NO在催化剂表面的吸附性能,进行了不同催化剂的NO-TPD测试(图7). NiAlO样品在160~240℃(α峰)和350~500℃(β峰)各有一个脱附峰. NiAlCeO催化剂两个脱附峰分别位于160~240℃和310~440℃.NO的脱附峰主要来自于亚硝酸盐的分解,而NO2的脱附峰主要由硝酸盐分解形成.值得注意的是低温区间NO的脱附峰强度随着Ce含量增加而增加,说明Ce的添加有助于催化剂表面亚硝酸盐物种的形成.同时,添加Ce后,高温区间的脱附峰脱附温度降低,这可能是由于部分硝酸盐更容易脱附.

2.6 原位红外吸附实验(In situ DRIFTS)
2.6.1 CO吸附 为了进一步研究CO-NO反应机理,进行了CO的原位红外吸附实验.图8是在不同温度下, CO吸附在NiAlCe20O催化剂表面的DRIFTS谱图.100℃在1650~1300cm-1区域的吸收峰主要是与碳酸盐型物种的形成有关[32].一般将波长大于2000cm-1的红外吸收峰归结于ν(C-O)的伸缩振动,即由线性吸附态的CO产生;当波长低于这个值时,则归属于桥式或孪生吸附态的CO[33].在NiAlCe20O上,并未出现这一系列吸收峰,表明CO在NiAlCe20O催化剂表面吸附较弱.而且该样品中,碳酸盐的红外吸收峰所处位置很高(红外波段在1643,1509和1400cm-1).碳酸盐主要是通过CO和与催化剂表面氧物种的反应形成,进一步表明催化剂具备较高的氧释放能力和流动性,这与已报道的含Ce固溶体具有高储氧能力一致.[34]当温度高于250℃时,碳酸盐开始分解,碳酸盐的吸收峰几乎完全消失,这是因为大部分CO与氧结合,生成CO2并释放.
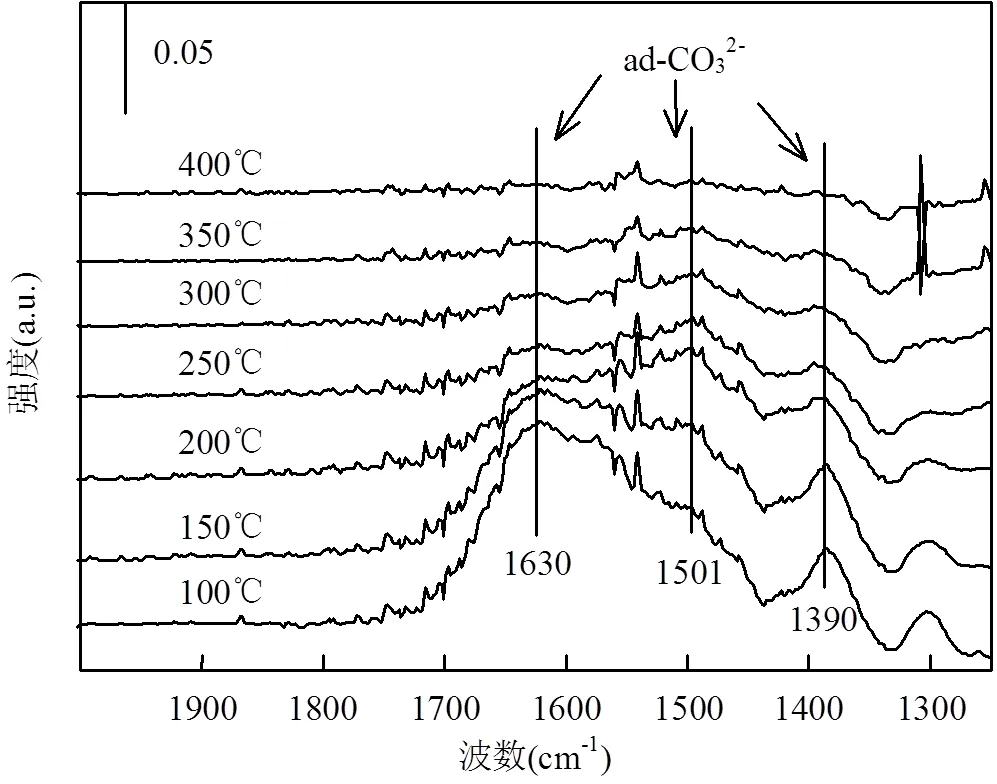
图8 CO/N2吸附于NiAlCe20O催化剂上的DRIFTS谱图
2.6.2 NO吸附 图9(a)是在不同温度下, NO吸附在NiAlO催化剂表面的DRIFTS谱图.在1560, 1531,1280,1240cm-1出现的峰值归属于吸附在NiO上的双齿硝酸盐[35],1615,1311cm-1处的峰归属于吸附在Al2O3上的桥式硝酸盐[35].以上结果表明对于NiAlO, NO吸附在催化剂表面主要形成硝酸盐物种.图9(b)是NO吸附在CeO2催化剂表面的原位红外谱图.在1199,1278cm-1处出现的强吸收峰说明存在吸附在Ce4+上的双齿亚硝酸盐物种(Ce4+=O2N)[36-37].随着温度的升高,该峰出现红移,表明在氧空位或者是其他缺陷位置,形成了双齿Ce+=O2N[38].在100~200℃时, 1547cm-1处的吸收峰归属于吸附在Ce上的单齿硝酸盐(Ce+=ONO2-)[39].当吸附温度在300~400℃时,由于螯合双齿硝酸盐(Cex+= O2NO-)的形成,在1523~1556,1224,1031,1008cm-1处出现了一系列吸收峰[36-37].此外,低温时,1617, 1570cm-1处出现的吸收峰可归属于气态或者弱吸附性态的NO2[40].这些数据表明,在CeO2上NO的吸附低温时主要以亚硝酸形式存在,随着吸附温度升高形成稳定的硝酸盐物种.

图9(c)是不同温度下NO吸附在NiAlCe20O催化剂表面的DRIFTS谱图.可以看出,在1240,1280, 1531,1560cm-1出现了双齿硝酸盐(非对称NO振动,ν3)的吸收峰[41-42].与前面的结果进行对比,在1199,1590cm-1的吸收峰可以分别归属于双齿亚硝酸盐物种和桥式硝酸盐物种.而在1452cm-1出现的吸收峰则归属于螯合亚硝酸盐物种[35,43].温度从100 ℃升高到250℃时,NO2和亚硝酸盐物种的吸收峰强度逐渐减弱,这与NO-TPD中催化剂在160~240℃的脱附峰相对应.当温度从300℃升高至400℃,1560,1531,1280,1240cm-1处的吸收峰峰强减弱,说明硝酸盐的吸附能力降低开始分解,这与NO-TPD中350~410℃的脱附峰相对应.
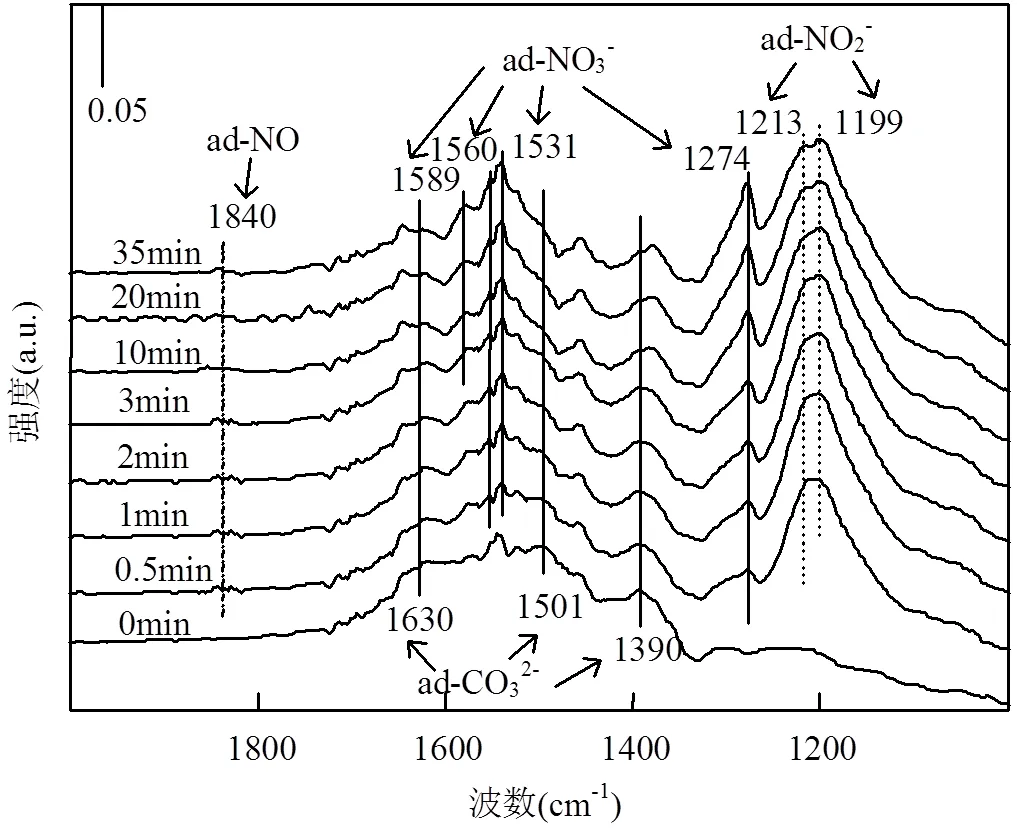
图10 NiAlCe20O催化剂吸附CO/N2 30min,经N2吹扫后通入NO/N2后表面的DRIFTS谱图

图11 NiAlCe20O催化剂吸附NO/N2 30min,经N2吹扫后通入500´10-6 CO/N2后表面的DRIFTS谱图
2.6.3 瞬态反应 图10是200℃下,NiAlCe20O先吸附CO后吸附NO的谱图.结果与NO和CO共吸附类似,稍有不同的是,实验过程中出现了硝酸盐(1274,1531,1560cm-1处)逐渐增加的现象,可能是由于没有连续通入CO,NO与表面氧反应生成硝酸盐,显然这不利于NO分解.
图11是在200℃下,NiAlCe20O先吸附NO后吸附CO的谱图,与200℃下NO-FTIR的结果相比(图9(c)),两者唯一的区别在于,该实验中出现了3个归属于碳酸盐的特征峰.

图12 CO-NO/N2在NiAlCe20O催化剂上吸附的DRIFTS谱
2.6.4 CO-NO共吸附 图12是温度为200℃时, NO和CO 在NiAlCe20O上的共吸附谱图.1390,1501, 1630cm-1处的吸收峰与碳酸盐的形成有关,这与CO吸附在NiAlCe20O的结果一致.结合CeO2和NiAlCe20O上的NO-DRIFTS结果可知,1199cm-1处的吸收峰与CeO2上的双齿亚硝酸盐有关.1213cm-1处的吸收峰可能是由吸附在NiO上的桥式亚硝酸盐物种造成[35].值得注意的是,在1840cm-1处出现了一个新的弱吸收峰,归属于吸附在NiO上的NO.这表明随着NO氧化程度降低,吸附态的NO开始出现,无疑有利于NO的分解反应.总之,与NO-FTIR结果进行对比,主要有两点不同:(1)吸附在NiO和CeO2上的亚硝酸盐吸收峰峰强显著增加.同时,出现了吸附态的NO,说明了NO氧化程度显著降低;(2)随着反应时间的增加,碳酸盐的吸收峰逐渐增强(1390, 1501,1630cm-1),而亚硝酸盐峰强逐渐减弱(1199, 1213cm-1),说明表面氧更倾向于与CO反应生成碳酸盐,而大多数通入的NO可能被还原为N2O或者N2.
结合以上的FTIR结果与已报导的结果[44],我们可以推断出NiAlCe20O上CO-NO的反应路径:(1)CO消耗表面氧来形成氧空位;(2)NO在氧空位上分解生成N2O或N2;(3)NO解离产生的氧留在氧空位上,随后被CO去除.
3 结论
3.1 NiAlCeO表现出比NiAlO更高的CO-NO反应活性
3.2 Ni进入Ce-O晶格形成Ni-Ce-O固溶体,由此产生的氧空位是反应的活性位点.
3.3 NiAlCeO上的CO-NO反应,同时存在2个反应:催化剂表面被CO还原并产生空位;NO被表面氧化成NO2,亚硝酸盐或硝酸盐.前者有利于CO-NO反应发生,而后者则相反.
[1] Twigg M V. Progress and future challenges in controlling automotive exhaust gas emissions [J]. Applied Catalysis B Environmental, 2007, 70(1-4):2-15.
[2] Roy S. Catalysis for NOabatement [J]. Applied Energy, 2009,86(11): 2283-2297.
[3] Yao X. Research progress on the catalytic elimination of atmospheric molecular contaminants over supported metal-oxide catalysts [J]. Catalysis Science & Technology, 2014,4(9):2814-2829.
[4] Tang C, Zhang H, Dong L.Ceria-based catalysts for low-temperature selective catalytic reduction of NO with NH3[J]. Catalysis Science & Technology, 2016,6(5):1248-1264.
[5] Chmielarz L. Catalytic activity of Co-Mg-Al, Cu-Mg-Al and Cu-Co-Mg-Al mixed oxides derived from hydrotalcites in SCR of NO with ammonia [J]. Applied Catalysis B Environmental, 2002,35(3): 195-210.
[6] Carja G. Mesoporous mixed oxides derived from pillared oxovanadates layered double hydroxides as new catalysts for the selective catalytic reduction of NO by NH3[J]. Applied Catalysis B Environmental, 2004,47(1):59-66.
[7] Basile F. Performance of new Pt and Pt-Cu on hydrotalcite-derived materials for NOstorage/reduction [J]. Topics in Catalysis, 2004, 30-31(1):223-227.
[8] Yu J J. Novel NO trapping catalysts derived from Co-Mg/X-Al (X=Fe, Mn, Zr, La) hydrotalcite-like compounds [J]. Environmental Science & Technology, 2015,41(4):1399-404.
[9] Li Q. Performance of K-promoted hydrotalcite-derived CoMgAlO catalysts used for soot combustion, NOstorage and simultaneous soot-NOremoval [J]. Applied Catalysis B Environmental, 2009, 91(1/2):406-415.
[10] 陈英红.水滑石类复合氧化物在CO+NO反应中的应用 [J]. 分子催化, 2000,14(4):270-274.
[11] Dai F F. Performance of Ce substituted hydrotalcite-derived mixed oxide catalysts Co2.5Mg0.5Al1-x%Ce%O used for soot combustion and simultaneous NO-soot removal [J]. Fuel Processing Technology, 2012,104(12):43-49.
[12] Palomares A E. Reactivity in the removal of SO2and NOon Co/Mg/Al mixed oxides derived from hydrotalcites [J]. Applied Catalysis B Environmental, 1999,20(4):257-266.
[13] Wang Y. Catalytic reduction of NO by CO over NiO/CeO2catalyst in stoichiometric NO/CO and NO/CO/O2reaction [J]. Applied Catalysis B: Environmental, 2008,81(1):141-149.
[14] Tang K. The Effect of Exposed Facets of Ceria to the Nickel Species in Nickel-Ceria Catalysts and Their Performance in a NO+CO Reaction [J]. ACS Applied Materials & Interfaces, 2015,7(48):26839- 26849.
[15] Venkataswamy P. Mn-doped Ceria Solid Solutions for CO Oxidation at Lower Temperatures [J]. Catalysis Letters, 2015,162(162):1-14.
[16] Kosacki I. Raman scattering and lattice defects in nanocrystalline CeO2thin films [J]. Solid State Ionics, 2002,149(1):99-105.
[17] Brussino P. NiCe/γ-Al2O3coated onto cordierite monoliths applied to Oxidative Dehydrogenation of Ethane (ODE) [J]. Catalysis Today, 2016,273:259-265.
[18] Lee Y. Raman analysis of mode softening in nanoparticle CeO(2-δ) and Au-CeO(2-δ) during CO oxidation [J]. Journal of the American Chemical Society, 2011,133(33):12952-12955.
[19] Chang S. Shape-dependent interplay between oxygen vacancies and Ag-CeO2interaction in Ag/CeO2catalysts and their influence on the catalytic activity [J]. Journal of Catalysis, 2012,293(9):195-204.
[20] Wu Z. Probing defect sites on CeO2nanocrystals with well-defined surface planes by Raman spectroscopy and O2adsorption [J]. Langmuir, 2010,26(21):16595-16606.
[21] Shi W. Densely populated mesopores in microcuboid CeO2crystal leading to a significant enhancement of catalytic activity [J]. Journal of Materials Chemistry A, 2012,1(3):728-734.
[22] Zhong S.NO oxidation over Ni-Co perovskite catalysts [J]. Chemical Engineering Journal, 2015,275:351-356.
[23] Zhang H L. Activity and thermal stability of Pt/Ce0.64Mn0.16R0.2O(R = Al, Zr, La, or Y) for soot and NO oxidation [J]. Fuel Processing Technology, 2015,137:38-47.
[24] Hattori H. Heterogeneous Basic Catalysis [J]. Chemical Reviews, 1995,95(3):537-558.
[25] Watanabe S. Characterization of Structural and Surface Properties of Nanocrystalline TiO2-CeO2Mixed Oxides by XRD, XPS, TPR, and TPD [J]. Journal of Physical Chemistry C, 2009,113(32):14249- 14257.
[26] Shen B. The effect of Ce-Zr on NH3-SCR activity over MnO(0.6)/Ce0.5Zr0.5O2at low temperature [J]. Chemical Engineering Journal, 2014,236(2):171-180.
[27] Wang X. Influence of transition metals (M = Co, Fe and Mn) on ordered mesoporous CuM/CeO2catalysts and applications in selective catalytic reduction of NOwith H2[J]. RSC Adv, 2015,5(77):63135- 63141.
[28] Liu J. Alkaline-assisted Ni nanocatalysts with largely enhanced low-temperature activity toward CO2methanation [J]. Catal Sci Technol, 2016,6(11):3976-3983.
[29] Zhou H. Preparation of NiCe Mixed Oxides for Catalytic Decomposition of N2O [J]. Industrial & Engineering Chemistry Research, 2013,52(12):4504-4509.
[30] Shan W. Reduction property and catalytic activity of Ce1-xNiO2mixed oxide catalysts for CH4oxidation [J]. Applied Catalysis A General, 2003,246(1):1-9.
[31] Zhao C. Understanding the impact of aluminum oxide binder on Ni/HZSM-5for phenol hydrodeoxygenation [J]. Applied Catalysis B Environmental, 2013,132-133(1):282-292.
[32] Morlanés N. Reaction mechanism of naphtha steam reforming on nickel-based catalysts, and FTIR spectroscopy with CO adsorption to elucidate real active sites [J]. International Journal of Hydrogen Energy, 2013,38(9):3588-3596.
[33] Hu C. Temperature-programmed FT-IR study of the adsorption of CO and co-adsorption of CO and H2on Ni/Al2O3[J]. Journal of Molecular Catalysis A Chemical, 1996,110(2):163-169.
[34] Natesakhawat S. Deactivation characteristics of lanthanide-promoted sol-gel Ni/Al2O3catalysts in propane steam reforming [J]. Journal of Catalysis, 2005,234(2):496-508.
[35] Pozdnyakov D. Infrared spectroscopic study of the chemisorption of nitric oxide and nitrogen dioxide on metal oxides [J]. Kinet Katal, 1973,14:760-766.
[36] Azambre B. Adsorption and Desorption of NOon Commercial Ceria-Zirconia (CeZr1-xO2) Mixed Oxides: A Combined TGA, TPD-MS, and DRIFTS study [J]. Journal of Physical Chemistry C, 2009,113(30):13287-13299.
[37] Martínezarias A. NO reaction at surface oxygen vacancies generated in cerium oxide [J]. Journal of the Chemical Society Faraday Transactions, 1995,91(11):1679-1687.
[38] Azambre B. Probing the Surface of Ceria-Zirconia Catalysts Using NOAdsorption/Desorption: A First Step Toward the Investigation of Crystallite Heterogeneity [J]. Journal of Physical Chemistry C, 2010,114(31):13300-13312.
[39] Qi G. Characterization and FTIR Studies of MnO-CeO2Catalyst for Low-Temperature Selective Catalytic Reduction of NO with NH3[J]. The Journal of Physical Chemistry B, 2004,108(40):15738-15747.
[40] Qi G.MnO-CeO2mixed oxides prepared by co-precipitation for selective catalytic reduction of NO with NH3at low temperatures [J]. Applied Catalysis B Environmental, 2004,51(2):93-106.
[41] Ohlsen J R. ChemInform Abstract: Characterization of asymmetric nitric oxide dimer O=N=O=N by resonance raman and infrared spectroscopy [J]. Chemischer Informationsdienst, 1979,10(4):6948- 6955.
[42] Hadjiivanov K. Identification of Neutral and Charged NOSurface Species by IR Spectroscopy [J]. Catalysis Reviews, 2000,42(1/2):71- 144.
[43] Centi G. Adsorption and reactivity of NO on copper-on-alumina catalysts: I. Formation of nitrate species and their influence on reactivity in NO and NH3conversion [J]. Journal of Catalysis, 1995,152(1):75-92.
[44] Wang Y. Catalytic reduction of NO by CO over NiO/CeO2catalyst in stoichiometric NO/CO and NO/CO/O2reaction [J]. Applied Catalysis B Environmental, 2008,81(1/2):141-149.
Study on Ce-doped Ni-Al-Ocatalysts for NO reduction by CO.
GUO Lei1, ZHANG Tao1, CHANG Hua-zhen1*, LI Jun-hua2
(1.School of Environment and Natural Resources, Renmin University of China, Beijing 100872, China;2.School of Environment, Tsinghua University, Beijing 100084, China)., 2018,38(9):3313~3321
A series of Ce-doped Ni-Al-Ocatalysts were prepared by a urea-hydrolysis method, and were applied for CO-NO reaction. The results showed that Ce-doped Ni-Al-Oexhibited much higher NO conversions than Ni-Al-O. Furthermore, the activity increased with the raising of Ce doping ratio. More than 95% NO could be converted at temperature as low as 250°C over NiAlCe30O and NiAlCe20O. These catalysts also showed excellent resistance to H2O. Ce-doped Ni-Al-Ocatalysts were characterized by XRD, N2physisorption, Raman, XPS, H2-TPR, NO-TPD, and in situ DRIFTs. XRD and Raman results showed that Ni-Ce-O solid solution was formed in Ni-Al-Ce-Ocatalyst. Meanwhile, oxygen vacancies increased with the increasing of Ce doping ratio. H2-TPR results indicated that the redox ability was improved by Ce doping. NO-TPD and in situ DRIFTs results revealed that the increasing number of oxygen vacancies resulted from Ce doping were beneficial to the dissociation of NO to N2or N2O rather than the oxidation of NO to nitrates.
Ni-Al-Ce-O;CO-NO;oxygen vacancy;Ce-doped
X505
A
1000-6923(2018)09-3313-09
郭 磊(1992-),男,湖南益阳人,中国人民大学硕士研究生,研究方向为大气污染控制工程.发表论文1篇.
2018-03-05
国家重点研发计划项目(2016YFC0203900,2016YFC0203901);国家自然科学基金项目(51778619,21577173)
* 责任作者, 副教授, chz@ruc.edu.cn
