Modern Antiplatelet Therapy: When ls Clopidogrel the Right Choice?
2018-09-17PunagDivanjiMDandKendrickShunkMDPhD
Punag Divanji, MD and Kendrick Shunk, MD, PhD
1The University of California San Francisco and The San Francisco Veteran Affairs Hospital, 505 Parnassus Ave, M1182,San Francisco, CA 94143-0124, USA
Abstract Platelet inhibition with aspirin is the cornerstone of medical therapy for coronary artery disease. In the era of percutaneous coronary intervention with drug-eluting stents, dual anti-platelet therapy with the addition of clopidogrel(Plavix©, Bristol-Myers Squibb, New York, NY) became the mainstay of therapy. However, with the advent of newer oral antiplatelet medications, including prasugrel (Effient©, Eli Lilly, Indianapolis, Indiana) and ticagrelor (Brilinta©,Astra-Zeneca, Cambridge, United Kingdom), choosing the appropriate platelet inhibitor has become more nuanced. The optimal regimen differs based on patient characteristics, clinical situation, and the condition being treated, with differing risk and safety profiles for these medications. This review explores the appropriate use of antiplatelet therapy for stable ischemic heart disease, acute coronary syndrome, stroke, and peripheral vascular disease. Furthermore, we evaluate the data behind the use of antiplatelet therapy in patients on oral anticoagulation. By understanding the biochemistry of platelet aggregation, the pharmacology of platelet inhibitors, and the extensive clinical trial data that informs current guideline recommendations we aim to better understand the role of clopidogrel in patients with cardiovascular disease.
Keywords: antiplatelet therapy; P2Y12 inhibitor; thienopyridine; acute coronary syndrome; stable coronary artery disease
lntroduction
Antiplatelet agents are protective in a variety of atherosclerotic cardiac and vascular diseases, including acute myocardial infarction (MI), ischemic stroke,non-ST elevation acute coronary syndrome (NSTEACS), stable angina, and peripheral artery disease(PAD). Although aspirin is the mainstay of antiplatelet therapy, an adjunctive second antiplatelet agent is often used to produce additional benefits in some clinical circumstances [1]. The oral antiplatelet drugs, including ticlopidine, clopidogrel, prasugrel, and ticagrelor, act on the platelet P2Y12receptor to inhibit platelet activation and aggregation. More than 600,000 patients received coronary stents in the United States in 2017, necessitating the use of antiplatelet agents [2]. Thus a thorough understanding of the various platelet inhibitors, their mechanism of action, and their indications is paramount.
The P2Y12 Receptor
Thienopyridines (ticlopidine, clopidogrel, and prasugrel) bind to the extracellular nucleotide P2Y12receptor, found on the cell surface of platelets, glial cells, and vascular smooth muscle cells, to inhibit platelet aggregation. When activated by its ligand,adenosine diphosphate (ADP), the Giprotein–coupled P2Y12receptor initiates an intracellular cascade that culminates in amplified platelet aggregation(Figure 1). In response to ADP binding, the Giprotein releases alpha and beta subunits that serve specific roles in the intracellular process [3]. The alpha subunit inhibits adenylyl cyclase, a protein responsible for the production of cyclic adenosine monophosphate (cAMP). Decreased cAMP production leads to decreased phosphorylation of vasodilator-stimulated phosphoprotein (VASP). The dephosphorylated form of VASP serves to activate the glycoprotein IIb/IIIa receptor, a key component of platelet aggregation.Simultaneously, the beta subunit activates phosphatidylinositol 3-kinase. This enzyme prompts conformational activation of integrin αIIbβ3, a cell adhesion molecule important in platelet aggregation[4]. Phosphatidylinositol 3-kinase also activates the serine/threonine protein kinase B and Rap1b guanosine triphosphate binding proteins, which, in turn,amplify the glycoprotein IIb/IIIa receptor to potentiate platelet aggregation. Furthermore, the beta subunit prompts release of platelet granules, which contain platelet agonists such as ADP, fibrinogen, factor V,and P-selectin [5]. Through this proinflammatory cascade, activation of the P2Y12receptor enhances platelet aggregation, prompts granule release, and increases the procoagulant/prothrombotic milieu.
Thienopyridines
Ticlopidine
Ticlopidine, the first compound of this class, came to the market in 1979 for stroke prevention. However,the medication is fraught with severe side effects,including gastrointestinal disturbances (vomiting,diarrhea, dyspepsia), rashes (purpura), hyponatremia,nephrotic syndrome, hepatotoxicity, neutropenia,and thrombocytopenia [6]. Since the introduction of newer agents, it has been largely supplanted by clopidogrel. As the drug is rarely used in modern clinical practice, it will not be discussed further in this review.
Clopidogrel
Pharmacology
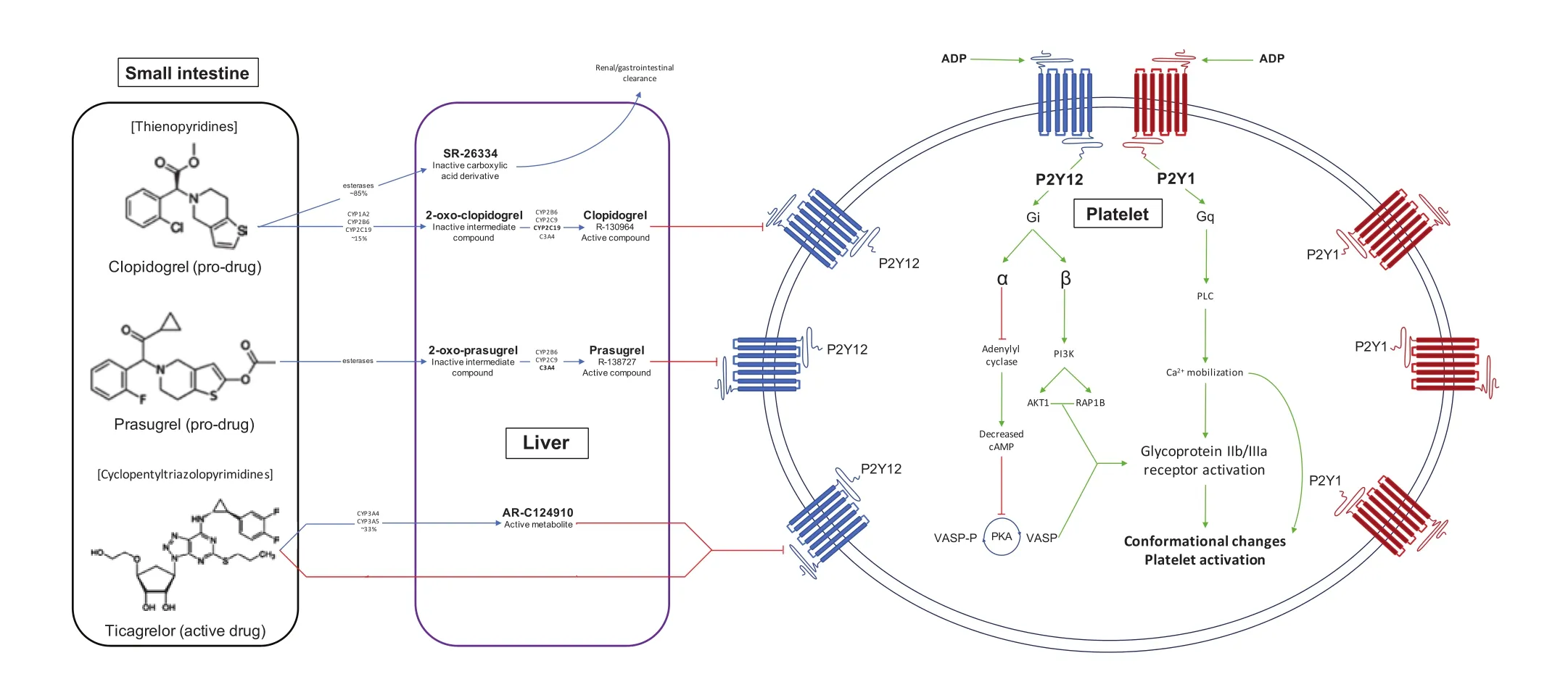
Figure 1 ADP-Receptor Antagonists Bind Irreversibly (Clopidogrel and Prasugrel) or Reversibly (Ticagrelor and Cangrelor)to the P2Y12 Receptor, Thereby Inhibiting Ca2+ Mobilization and Activation of the GPIIb/IIIa Receptor.Inhibition of Ca2+ mobilization reduces secretion of pro-aggregetory substances and activation of the GPIIb/IIIa receptor, preventing conformational changes that result in platelet aggregation.
Introduced in the United States in 1998, the oral P2Y12inhibitor clopidogrel bisulfate (Figure 1), a thienopyridine, is ingested as an inactive prodrug.Approximately 50% of this compound is absorbed in the gastrointestinal tract by the drug efflux transporter P-glycoprotein (encoded by theABCB1gene). The inactive metabolite is then delivered to the liver for further processing. In the liver, up to 85% of the delivered clopidogrel is hydrolyzed during first-pass metabolism by carboxylesterase 1 [7].This converts the prodrug into an inactive carboxylic acid derivative that is excreted mainly via the renal (50%) and gastrointestinal (46%) systems. The remaining clopidogrel is metabolized via a group of cytochrome 450 enzymes (CYP) into its active form in a two-step process. In the first step, CYP1A2,CYP2B6, and CYP2C19 convert clopidogrel into 2-oxoclopidogrel, a still inactive intermediate compound. This compound is then converted to the clopidogrel active metabolite (R-130964) by CYP2B6,CYP2C9, CYP2C19, and CYP3A4. CYP2C19 is the most active enzyme of this group, accounting for approximately 50% of the clopidogrel active metabolite that is produced [8]. Importantly, both 2-oxoclopidogrel and the active metabolite are hydrolyzed by carboxylesterase 1, such that approximately 2%of the ingested prodrug form of clopidogrel enters the systemic circulation in the active form. The prodrug form of clopidogrel has a half-life of approximately 7–8 h, while the active metabolite has a half-life of only 30 min; however, because of its irreversible binding to the P2Y12receptor, the drug persist for the lifespan of the affected platelet, approximately 7 days [9]. Most (>95%) of this circulating clopidogrel, including both the active metabolite and the inactive metabolite, is protein bound. Thus any factors affecting protein binding, including the relative concentration of active/inactive metabolites, pH, and albumin concentration, can affect the bioavailability of active clopidogrel. Furthermore, the extensive hepatic metabolism of clopidogrel by the inducible CYP system leads to many potential drug-drug interactions. The active metabolite binds irreversibly [10]to the P2Y12receptor by forming disulfide bonds [11]with cysteine residues at the ADP-binding site, leaving the receptor nonfunctional for the remainder of the platelet lifespan (up to 9 days) [12]. In addition to direct platelet inhibition, clopidogrel also decreases the levels of markers of inflammation, with decreased production of C-reactive protein, CD40 ligand, and P-selectin (CD62) [13].
Of the important attributes of antiplatelet agents,the time from administration to effective platelet inhibition is paramount. In patients with acute coronary syndrome (ACS), particularly those with ST elevation MI (STEMI), the time required to achieve adequate platelet inhibition is crucial to prevent complications such as early drug-eluting stent thrombosis. Platelet inhibition by clopidogrel is dose and time dependent; when clopidogrel is administered as a standard 75 mg dose, the time to initial activity of the active metabolite is 2–4 h,achieving a peak effect as late as 3–7 days [14]. In practice, clopidogrel is typically administered with a loading dose of either 300 or 600 mg to achieve platelet inhibition as rapidly as possible. Assessment of platelet reactivity by means of the maximal intensity of ADP-induced platelet aggregation suggests a peak effect at 3–5 h with a dose of 300 mg [14].After the administration of 600 mg, the peak effect occurs at 2–3 h [15]. The dose-effect relationship with clopidogrel is nonlinear, with no incremental benefit to loading doses greater than 600 mg [16].Studies in patients undergoing percutaneous coronary intervention (PCI) for stable coronary artery disease (CAD) or ACS (both non–ST elevation MI[NSTEMI] and STEMI) had reduced major adverse cardiovascular events and infarct size with a loading dose of 600 mg, driven primarily by a decrease in stent thrombosis. However, there was a signal for increased major bleeding with high-dose clopidogrel [17–23].
Pharmacogenetics
The maximum effect of clopidogrel appears to be 50–60% platelet inhibition, although the individual’s response differs on the basis of a number of patient-specific factors. Up to 30% of white patients and 50% of Asian patients have an attenuated response to clopidogrel administration (with higher rates in people with diabetes), resulting in high on-treatment platelet reactivity, a phenomenon dubbedclopidogrel resistance[3, 24–26]. Platelet activity can be assessed by a number of different methods, although the gold standard is light transmittance aggregometry in response to ADP [27,28]. By aggregometry, an appropriate response to clopidogrel administration is defined as a more than 20% decrease in maximal platelet aggregation after administration of the P2Y12agonist ADP.Clopidogrel resistance, then, is defined in the laboratory as a decrease in maximal aggregation of 10%or more from the baseline [29]. However, because of the cumbersome nature of this test, the more commonly used methods involve bedside evaluation with commercially available tests: flow-cytometric analysis of VASP; the PFA-100®analyzer (Siemens Healthcare Diagnostics, Deerfield, IL, United States) looks at platelet aggregation in response to collagen/epinephrine or collagen/ADP cartridges under high-shear conditions; the VerifyNow®P2Y12test (Accumetrics, San Diego, CA, USA) looks at platelet aggregation of fibrinogen-coated beads in response to ADP [30–32]. With these tests, clopidogrel resistance is defined as platelet reactivity index greater than 50% by analysis of VASP phosphorylation and greater than 240 P2Y12reaction units by the VerifyNow assay [33]. Importantly, laboratory evidence of decreased clopidogrel response does not necessarily translate to clinical treatment failure; in practice, clopidogrel resistance is defined as recurrent thrombotic events despite appropriate clopidogrel therapy, suggesting high on-treatment platelet activity.
There are several potential patient-specific mechanisms for decreased platelet inhibition in response to clopidogrel, including drug-drug interactions,variable absorption/metabolism of the prodrug,alternative pathways for platelet activation, and genetic polymorphisms [34].
Clopidogrel and prasugrel (a third-generation thienopyridine) are subject to intestinal efflux by the ATP-dependent P-glycoprotein pump (also known asmultidrug resistance protein 1), encoded by theABCB1gene [35]. P-glycoprotein [1] limits the oral bioavailability of these drugs via control of intestinal absorption. Accordingly, individuals with the C3435 TT genotype, which results in impaired function of the intestinal drug-efflux transporter[36, 37], have lower circulating level of the active metabolite, with the potential for increased risk of major adverse cardiovascular events [38], although this remains controversial [39, 40].
Furthermore, the CYP system is susceptible to genetic polymorphisms that affect thienopyridine metabolism, with more than 48CYP2C19allelic variants described, ranging from null to increased enzymatic activity [41]. The two most common genetic variants responsible for decreased platelet inhibition are loss of function (LoF) in theCYP2C19andCYP3A4/CYP3A5genes, resulting in so-called poor metabolizers [3, 42]. As noted earlier, CYP2C19 is active in both of the sequential steps required to convert clopidogrel into its active metabolite. Reduced CYP2C19 activity results in shunting of clopidogrel toward esterification,increasing production of the inactive metabolite.Thus LoF results in a 32% reduction in the active metabolite and 9% reduction in maximal platelet aggregation [36, 43]. Together, the LoFCYP2C19polymorphisms cause 12% of the variation of platelet inhibition seen in patients receiving clopidogrel[44]. In this setting, the specificCYP2C19*2mutation accounts for most (>90%) of those with the reduced function allele [45]. This allele is present in 15% of white and black patients, and up to 35% of Asians, suggesting a large potential clinical impact[46].CYP2C19LoF mutations are associated with increased death from cardiovascular causes, nonfatal MI, and nonfatal stroke [26, 45, 47–49]. The risk associated with LoFCYP2C19variants increases significantly in patients undergoing PCI, who have an almost three-fold increase in stent thrombosis[43, 50, 51].
The most common polymorphism responsible for a gain of function is theCYP2C19*17variant,which is associated with enhanced enzyme activity [52, 53]. These so-called fast metabolizers have greater reduction in platelet aggregation and a resultant decrease in potential thrombotic events,but at the cost of increased bleeding [36, 54].
Other mutations in the CYP 450 system have been postulated to contribute to clopidogrel resistance, with CYP3A4 inhibitors leading to decreased production of the clopidogrel active metabolite [55].However, studies do not support a clinically significant role for these mutations in causing adverse cardiovascular events [56], although inducers of CYP3A4 may potentiate active metabolite formation, resulting in increased efficacy, but, again, at the cost of increased bleeding [55].
More than 50% of the population is affected by eitherABCB1orCYP2C19polymorphisms, raising concern for the potential risk of major adverse cardiovascular events due to high platelet reactivity while patients are receiving thienopyridines [4, 38].Because of the risk for variable platelet inhibition and potential adverse clinical outcomes, platelet function testing to guide the choice of P2Y12inhibitor has a class IIb recommendation for selected, high-risk patients undergoing PCI [57–59]. Otherwise, current guidelines note that the routine use of platelet function and genetic testing is not recommended (class III: no benefit) [57]. For these reasons, the black box warning on the package insert for clopidogrel warns that “patients homozygous for nonfunctional alleles of CYP2C19 gene (CYP2C19 poor metabolizers)formed less active metabolite and had reduced antiplatelet activity at recommended doses; CYP2C19 genotype tests are available to identify CYP2C19 poor metabolizers; consider using other platelet P2Y12 inhibitor treatment in CYP2C19 poor metabolizers” [60]. However, it is important to remember that not all cases of laboratory resistance translate to clinical resistance.
Drug-Drug Interactions
Ingested as a prodrug, clopidogrel must undergo enzymatic conversion to its active form, requiring the complex pathway described earlier. Each of these steps is susceptible to interaction with commonly used medications, both cardiac and noncardiac. The CYP system is particularly susceptible to inducers and inhibitors of its function, with an extensive list of agents that can affect the clinical response to thienopyridines (Table 1). The most commonly used drugs with the potential to inhibit the effects of clopidogrel include proton pump inhibitors (PPIs),3-hydroxy-3-methylglutaryl coenzyme A reductase(HMG-CoA reductase) inhibitors (statins), and calcium channel blockers (CCBs).
Proton Pump Inhibitors
To reduce the risk of gastrointestinal bleeding in patients receiving antiplatelet therapy, PPIs are often prescribed. Omeprazole, the most widely prescribed PPI, is ingested as a prodrug, requiring activation by CYP2C19 and CYP3A4/CYP3A5,similarly to clopidogrel [62]. Of the PPIs, omeprazole has a greater affinity for CYP2C19 than for CYP3A4/CYP3A 5 than other PPIs, suggesting a greater potential for interaction with clopidogrel and the potential safety of medications such as pantoprazole or esomeprazole [63].Accordingly, pharmacodynamic studies demonstrated decreased effectiveness of clopidogrel, but this finding was not consistent with the findings of observational studies [64]. However, a recent meta-analysis evaluating studies since 2012 suggested increased rates of major adverse cardiac events following PCI in patients taking omeprazole (but not pantoprazole) [65]. In the Clopidogrel and the Optimization of Gastrointestinal Events Trial (COGENT), the only randomized, doubleblind study to evaluate the cardiovascular implications of combining clopidogrel and omeprazole,patients treated with this combination had a lower incidence of gastrointestinal bleeding, with an incidence of cardiovascular events similar to that with clopidogrel and placebo [66]. However, there were several limitations to this study, including premature termination, smaller than expected number of events, wide confidence intervals, and small percentage of patients with theCYP2C19homozygous mutation, and definitive conclusions about this interaction could not be made [66].In light of these limitations, the Food and Drug Administration affirmed the warning on the clopidogrel label that concomitant use of clopidogrel and omeprazole should be avoided [67]. Further randomized controlled trials are needed to elucidate this issue.
HMG-CoA Reductase Inhibitors
To mitigate the risk of atherosclerotic cardiovascular events, statin medications, which inhibit HMG-CoA reductase so as to reduce endogenous cholesterol production, are commonly prescribed to patients with CAD or PAD. Several statins, including atorvastatin, simvastatin, lovastatin, and fluvastatin, are metabolized by CYP3A4/CYP3A5, and can theoretically interact with the metabolism of clopidogrel [68]. In 2003, Lau et al. [69] studied a potential interaction between atorvastatin and clopidogrel, and they concluded that competitive inhibition of CYP3A4/CYP3A5 by atorvastatin decreased the activation of clopidogrel, resulting in decreased platelet inhibition. However, several subsequent studies evaluated the interaction of the statins and clopidogrel, revealing no significant change in platelet inhibition or cardiovascular outcomes,especially when the higher (600 mg) loading dose is used [70–76]. For this reason, there are currently no contraindications to statin use in patients taking clopidogrel (Table 2).
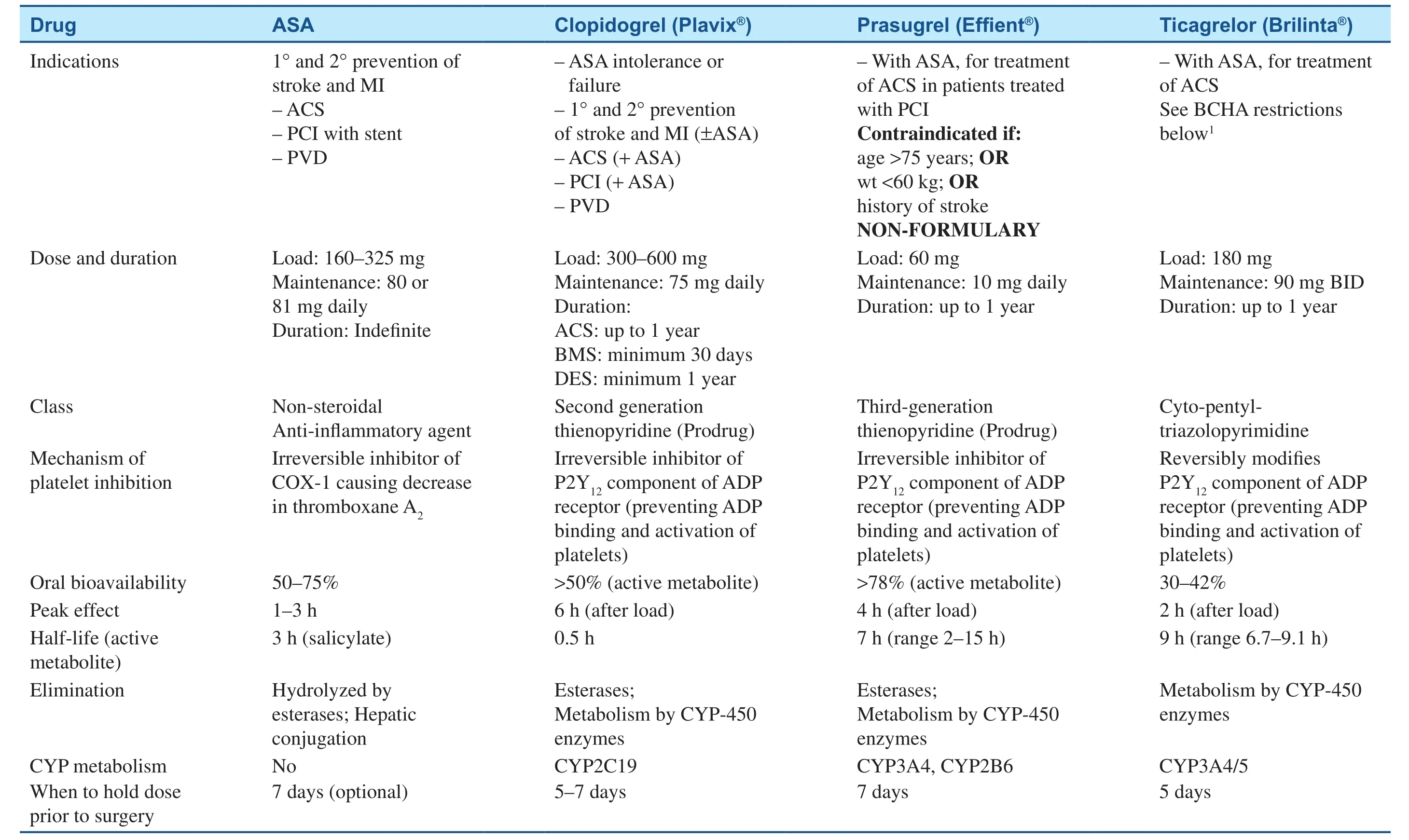
Table 1 Antiplatelet Drug Comparison Chart.
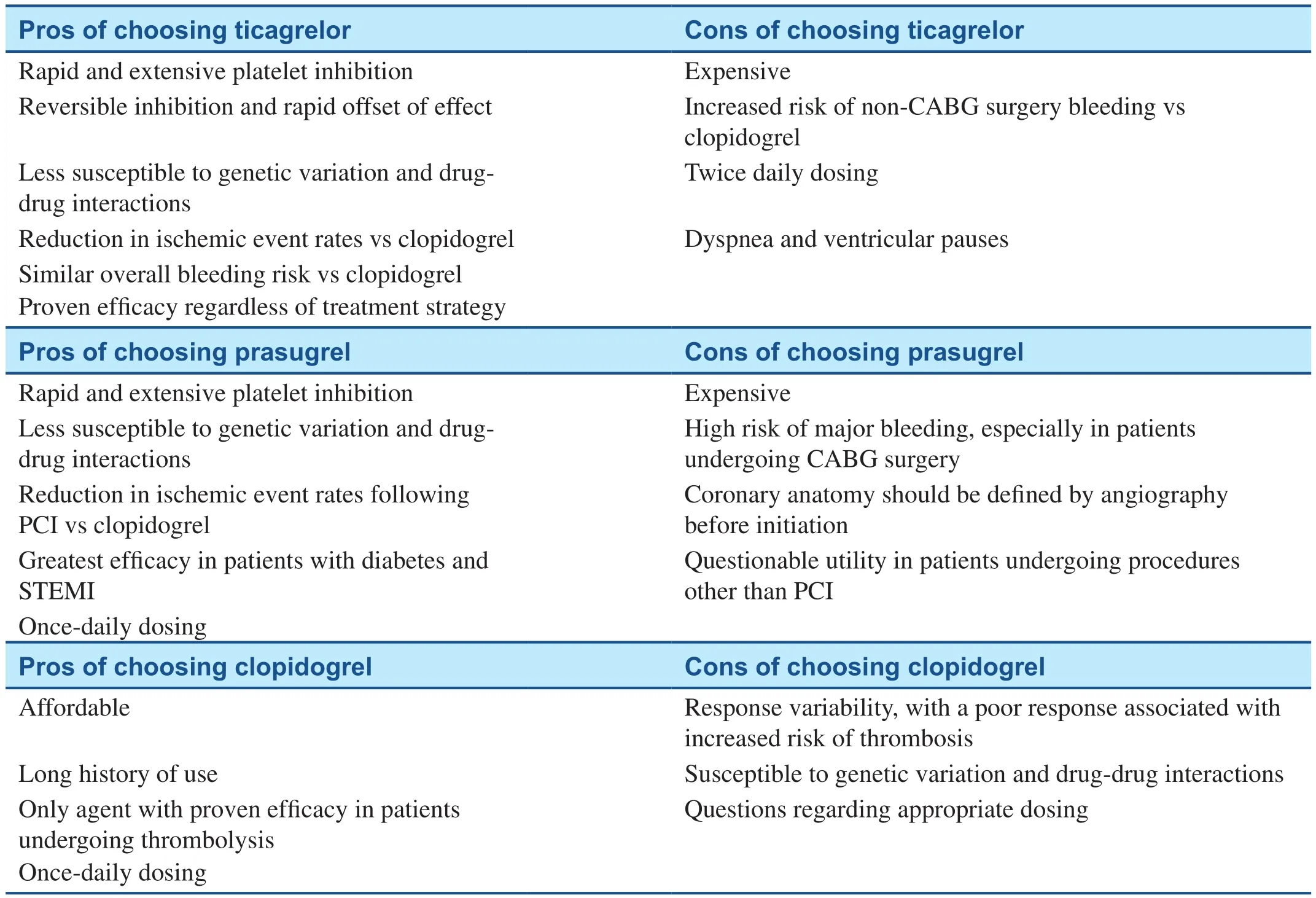
Table 2 Advantages and Disadvantages of the P2Y12 Receptor Inhibitors.
Calcium Channel Blockers
Frequently used in the management of hypertension, atrial fibrillation, and angina, CCBs can inhibit CYP3A4/CYP3A5, resulting in decreased concentration of the clopidogrel active metabolite and subsequent high on-treatment platelet activity [78].Many CCBs also inhibit P-glycoprotein, thereby decreasing clopidogrel efflux and resulting in higher circulating levels of the drug, attenuating the effect of CYP3A4/CYP3A5 inhibition. However, amlodipine, which does not inhibit P-glycoprotein, results in both decreased clopidogrel activation and persistent intestinal efflux, decreasing bioavailability of the active form [79]. The dihydropyridine class of CCBs have been implicated in this interaction, with several studies suggesting decreased platelet inhibition in patients receiving clopidogrel and concomitant CCBs [80–82]. However, there are conflicting recent studies regarding the clinical impact of CCBs(including amlodipine) on clopidogrel metabolism,with no consensus reached thus far [83–85]. Thus there are no significant restrictions on the use of CCBs with clopidogrel.
Azoles
The azole antifungal drugs, used for both treatment and prophylaxis of fungal infections in immunosuppressed patients, such as organ transplant recipients or patients with human immunodeficiency virus.These medications inhibit the CYP3A4/CYP3A5 isozyme, particularly ketoconazole, although the more commonly used medications are fluconazole and itraconazole [86]. Administration of ketoconazole leads to impaired platelet inhibition in patients taking clopidogrel, although there are no descriptions of clinically significant clopidogrel resistance in patients taking azole antifungals [87].
Clinical Outcomes
Coronary Artery Disease
The clinical efficacy of clopidogrel was first established by the Clopidogrel versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) trial in 1996,which evaluated 19,195 patients with recent stroke,or established PAD [88]. In this study, clopidogrel monotherapy versus aspirin monotherapy was associated with a significant 8.7% relative risk reduction compared with aspirin for the primary end point of ischemic stroke, MI, or vascular death (5.32% per year compared with 5.83% per year; P = 0.043).However, this benefit was most pronounced in the PAD subgroup, while the benefit in the stroke subgroup was not statistically significant; risks of hemorrhage were similar between the groups. This was followed by a comparison of clopidogrel versus ticlopidine in patients with ACS or patients undergoing PCI, demonstrating comparable efficacy and a better safety profile, including less bleeding, neutropenia, and thrombotic thrombocytopenic purpura [89–91]. The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) study firmly established the role of aspirin plus clopidogrel in the medical management of ACS, while the PCI substudy supported clopidogrel pretreatment followed by long-term therapy, each showing a reduction in major cardiovascular events at the cost of increased bleeding [92, 93]. Several subsequent trials furthered the role of long-term aspirin plus clopidogrel therapy after ACS, demonstrating a reduction in the relative risk of death, MI, and stroke after PCI [94,95] or fibrinolysis [96]. In the CHARISMA trial, the use of dual antiplatelet therapy (DAPT) including aspirin and clopidogrel did not, however, show significant benefit in a broad population of patients at high-risk of cardiovascular events, including those with stable CAD [97]. However, post hoc analysis revealed a signal for benefit in patients with symptomatic atherothrombotic disease, at the cost of increased bleeding.
With the efficacy of this clopidogrel established in ACS and stroke, trials began to focus on the optimal timing of clopidogrel administration and dose of clopidogrel. In patients undergoing PCI, a double dose regimen, including a 600 mg loading dose of clopidogrel followed by 150 mg for 1 week,reduced cardiovascular events and stent thrombosis when compared with the standard dose (300 mg load, 75 mg daily) [23]. However, extension of this double dose out to 6 months did not improve outcomes further [98]. The Atorvastatin for Reduction of Myocardial Damage during Angioplasty(ARMYDA)-2 trial investigated the dose-time relationship for clopidogrel in patients with stable CAD or NSTE-ACS about to undergo PCI. A significant decrease in major adverse cardiovascular events was discovered with the higher loading dose(600 mg) without an increase in major bleeding.Subsequent ARMYDA trials concluded that patients receiving long-term clopidogrel therapy with stable angina could safely undergo PCI without a reload,that patients receiving long-term clopidogrel therapy with ACS undergoing PCI had better outcomes with a 600 mg reload, and that giving the clopidogrel loading dose “in the lab” just before PCI was as safe and effective as a preload (6 h before PCI)[99–101]. In STEMI patients, pretreatment with the 600 mg clopidogrel loading dose before primary PCI was associated with a reduction in infarct size and 30 day major adverse cardiovascular events,while the 150 mg maintenance dose caused more potent platelet inhibition and decreased inflammation [21, 102]. In this setting, in patients with ACS,the current American College of Cardiology (ACC)/American Hear Association (AHA) guidelines recommend a clopidogrel 300 mg bolus and 75 mg maintenance dose for 1 year if ACS is managed medically or with fibrinolytics, versus a 600 mg bolus and 75 mg maintenance dose for 1 year if it is managed with PCI [103].
Ischemic Stroke
Building on the results of the CAPRIE trial, multiple trials evaluated the efficacy and safety of clopidogrel in noncardioembolic stroke or transient ischemic attack (TIA), demonstrating that clopidogrel and aspirin were superior to aspirin alone if used early (<24 h to 21 days) in ischemic stroke,but with an increased risk of bleeding [104]. This confirms the findings of previous studies looking at ischemic stroke in patients with symptomatic carotid disease [105–107]. However, in the CHARISMA trial, aspirin plus clopidogrel did not reduce the risk of the composite primary end point (MI, stroke of any cause, or death from cardiovascular causes)compared with aspirin alone, and the risk of bleeding was higher [97]. Subsequent trials confirmed the benefit from upfront DAPT using aspirin and clopidogrel, with no benefit and potential increased harm with long-term use [108, 109]. The role of DAPT in patients with ischemic stroke will be further evaluated in the upcoming Platelet-Oriented Inhibition in New TIA and Minor Ischemic Stroke (POINT)trial, a randomized, double-blind, multicenter clinical trial to determine whether clopidogrel at 75 mg/day (after a loading dose of 600 mg) is effective in improving survival free from major ischemic vascular events when treatment is initiated within 12 h in patients receiving aspirin [110]. Thus current guidelines offer a class IIb recommendation for the use of clopidogrel at 75 mg/day with aspirin for up to 90 days in patients with recent stroke or TIA(within 30 days) attributable to severe stenosis (70–99%) of a major intracranial artery, with the caveat that long-term use of DAPT with clopidogrel plus aspirin (i.e., >90 days) is not recommended because of the increased risk of bleeding and all-cause mortality, unless there is another indication (e.g., coronary stent). Furthermore, the guidelines offer a class IIa recommendation for clopidogrel monotherapy in place of aspirin monotherapy [111].
Peripheral Artery Disease
PAD is an independent risk factor for CAD, and both entities have atherothrombotic complications[112]. Management strategies include medical therapy, balloon angioplasty, and stent placement,with various strategies regarding the management of antiplatelet therapy. In the MATCH trial (aspirin and clopidogrel compared with clopidogrel alone after recent ischemic stroke or TIA in highrisk patients), PAD patients in the high-risk category derived significant cardiovascular risk benefit with DAPT with clopidogrel [108]. However, when clopidogrel therapy was added to aspirin therapy in PAD patients in the CHARISMA trial, they did not have significant improvement in the primary efficacy end point of MI, stroke, or vascular death,although those at highest risk (i.e., established PAD)did see a decrease in the rate of hospitalization for ischemic events, including MI [97, 113]. In the aforementioned CAPRIE trial, the benefit of clopidogrel over aspirin in terms of cardiovascular risk reduction was driven predominantly by the PAD subgroup [88]. On the basis of these results, the ACC/AHA guidelines offer a class I recommendation for the use of clopidogrel as a safe and effective alternative antiplatelet therapy to aspirin to reduce the risk of MI, ischemic stroke, or vascular death in individuals with symptomatic atherosclerotic lower extremity PAD. Even in asymptomatic individuals,antiplatelet therapy has a class IIa indication in this group [114].
Atrial Fibrillation
In the population of patients with an indication for DAPT and anticoagulation (i.e., CAD and atrial fibrillation), triple therapy with aspirin, clopidogrel, and warfarin incurs an up to threefold increase in bleeding risk, which portends worse prognosis and predicts increased mortality in patients after PCI [115–117]. There is potentially a need for triple therapy in at least 5% of patients undergoing PCI, and the incidence of atrial fibrillation is expected to increase in the coming years[103]. The WOEST trial (use of clopidogrel with or without aspirin in patients taking oral anticoagulant therapy and undergoing PCI) evaluated triple therapy with DAPT and warfarin versus clopidogrel monotherapy and warfarin in patients undergoing PCI [118]. This small trial of 563 patients found that the warfarin plus clopidogrel alone (double therapy) was associated with a significant reduction in bleeding complications compared with warfarin plus DAPT (triple therapy),without an increase in thrombotic complications.The findings of this study were further bolstered by registry data [119, 120]. However, the most recent ACC/AHA NSTE-ACS guidelines continue to recommend triple therapy in patients with NSTE-ACS [121], although the 2014 atrial fibrillation guidelines offer a class IIb recommendation for double therapy in this setting [122]. With the advent of direct oral anticoagulants (dabigatran,rivaroxaban, apixaban, edoxaban) and their burgeoning use in clinical practice, the question of anticoagulation in patients with atrial fibrillation undergoing PCI must be considered in this context.Akin to the WOEST trial, investigators looked at rivaroxaban plus P2Y12inhibitor therapy versus traditional triple therapy in atrial fibrillation patients undergoing coronary revascularization,discovering that a rivaroxaban-based strategy was associated with a lower frequency of clinically significant bleeding, all-cause death, and rehospitalization, with similar rates of major adverse cardiac events, including stent thrombosis [123].Similarly, investigators evaluated double therapy with dabigatran and clopidogrel versus traditional triple therapy, finding that dual therapy with dabigatran was superior to triple therapy with warfarin in preventing bleeding, without an increase in the risk of death, MI, or stroke [124]. On the basis of the findings of these trials, the 2017 European Society of Cardiology guidelines recommend dual therapy with an oral anticoagulant after 1 month of triple therapy in patients with high bleeding risk,adding that direct oral anticoagulants may be considered in place of warfarin [125]. Importantly, all patients in the WOEST trail, more than 90% of patients in the PIONEER AF-PCI trail, and more than 80% of patients in the RE-DUAL PCI trial were taking clopidogrel as the P2Y12inhibitor of choice, suggesting a significant role for clopidogrel in patients with atrial fibrillation undergoing PCI [118, 123, 124].
After Transcatheter Aortic Valve Replacement
Antiplatelet therapy has been a cornerstone of postprocedure management for patients undergoing transcatheter aortic valve replacement (TAVR) since the early clinical trials. In the United States, current practice involves upfront DAPT with clopidogrel at a dose of 75 mg for 6 months with balloon-expandable valves and for 3 months with self-expanding valves, along with aspirin at a dose of 75–100 mg lifelong [126]. This recommendation arises from the original TAVR approval data, likely due to the standard clinical practice for other endovascular stents [127–132]. To better evaluate the role of antiplatelet therapy after TAVR, the Aspirin Versus Aspirin + Clopidogrel following Transcatheter Aortic Valve Implantation (ARTE) trial randomized 222 patients presenting for TAVR with the Edwards balloon-expandable valve to receive either aspirin alone or aspirin plus clopidogrel following the TAVR procedure. At 3 months, those receiving DAPT were more likely to experience a primary end point event (death, MI, stroke/TIA, or major/life-threatening bleeding) than those receiving aspirin alone (15.3 vs. 7.2%; P = 0.065), driven primarily by increased major and minor bleeding[133]. This is particularly important in the setting of recent data suggesting rates of subclinical leaflet thrombosis approaching 15%, with no significant difference between DAPT and single antiplatelet therapy [134, 135]. Furthermore, severer, clinically manifest leaflet thrombosis may be associated with stroke, cardiogenic shock, and death [136], emphasizing the need for further research. To better understand the role of antiplatelet therapy after TAVR,two larger-scale trials, POPular-TAVI and CLOE,will evaluate aspirin therapy alone versus aspirin and clopidogrel therapy in the first 3 months after TAVR [137]. Notably, anticoagulation therapy appears to more effectively prevent leaflet thrombosis in comparison with antithrombotic therapy,an issue that will be addressed by the upcoming AUREA (vitamin K antagonist), GALILEO (rivaroxaban), and ATLANTIS (apixaban) trials.
Triple Antiplatelet Therapy
Cilostazol is a selective inhibitor of phosphodiesterase type 3 that results in reversible inhibition of platelet aggregation, in addition to vasodilatory and antiproliferative effects [138]. The concept of triple antiplatelet therapy with aspirin, clopidogrel, and cilostazol was intended to bolster antiplatelet activity. This has been studied in the context of both CAD and PAD, with evidence to suggest that the addition of cilostazol to standard therapy with aspirin and clopidogrel significantly decreases platelet activity when compared to standard therapy alone [139–141]. Initial work suggested that triple antiplatelet therapy with these agents may decrease the rates of stent thrombosis and target vessel failure without an increased risk of bleeding, both in CAD [141, 142]and in PAD [143–145]. However, recent trial data suggest no clinical benefit with regard to the incidence of cardiac death, nonfatal MI, or stroke, even in complex PCI [140, 146, 147]. More research is needed in this area to better adjudicate the role of cilostazol in addition to aspirin and clopidogrel.
Thus clopidogrel is used as an effective antiplatelet agent in a variety of situations, including ACS, stable CAD, PAD, stroke, after TAVR, and as double therapy for atrial fibrillation patients undergoing PCI. However, the extensive metabolism required for activation potentially limits its efficacy in patients with common genetic polymorphisms(CYP2C19,ABCB1), and drug-drug interactions may play a role, although this requires further investigation. Nonetheless, for the majority of patients requiring antiplatelet therapy beyond aspirin, clopidogrel can be considered a safe first option.
Prasugrel
Pharmacology
Like clopidogrel, the third-generation thienopyridine prasugrel irreversibly binds the P2Y12receptor and begins as a prodrug that requires transformation to its active form [148]. Prasugrel is administered at an oral loading dose of 60 mg, followed by daily maintenance doses of 10 mg, with the consideration of a 5 mg dose in those weighing less than 60 lb (27 kg) [149]. More than 80% of the prodrug is absorbed in the small intestine, where carboxylesterase 2 hydrolyzes the prodrug to 2-oxoprasugrel [150, 151]. The intermediate compound is then converted to the prasugrel active metabolite in the liver by the CYP system [152]. As opposed to the two-step CYP metabolism of clopidogrel involving CYP2C19, prasugrel is metabolized primarily by CYP3A4/CYP3A5 in one step, making it less susceptible to inactivation and interference from CYP inhibitors [153]. CYP3A4/CYP3A5 accounts of up to 70% of the conversion of 2-oxoprasugrel to the prasugrel active metabolite, with CYP2B6 accounting for up to 26% [154]. Prasugrel metabolism is highly efficient during both absorption and conversion to the active metabolite; the active metabolite is detected within 15 min, the mean time to peak plasma concentration is approximately 30 min, and the median plasma half-life is approximately 7.4 h [155, 156]. The drug is excreted mostly via the renal (70%) and gastrointestinal (30%) systems [87]. Prasugrel reaches higher concentrations of its active metabolite than clopidogrel, with the highest concentration typically found in Asian patients [157]. Given this rapid and efficient activation, prasugrel is tenfold more potent than clopidogrel [158], yielding prompt, irreversible platelet inhibition by forming a disulfide bridge with a free cysteine residue on the P2Y12receptor [53]. Thus prasugrel administration results in faster, more consistent, and more potent inhibition of platelet aggregation when compared with clopidogrel [159, 160].
Pharmacogenetics
The extensive metabolism of prasugrel via CYP3A4 obviates the issue with incomplete response due toCYP2C19mutations, as this isozyme accounts for only a minority of active metabolite production [151, 161]. Additionally, prasugrel does not have a significant inactivation pathway, resulting in consistent plasma levels of the active metabolite[162]. Accordingly, on-treatment platelet reactivity is much lower for prasugrel when compared with clopidogrel, with a significantly lower incidence of laboratory hyporesponsiveness to the third-generation drug [163]. The aforementioned common functional CYP genetic variants (such asCYP2C19*2)do not affect the prasugrel active metabolite levels, platelet inhibition, or clinical cardiovascular events [164, 165]. As previously mentioned,prasugrel is also subject to intestinal efflux by the ATP-dependent P-glycoprotein pump, encoded by theABCB1gene. Unlike clopidogrel, however,theABCB13435 TT genotype was not associated with impaired platelet inhibition of adverse clinical outcomes in patients taking prasugrel [38].Furthermore, in cases of clinical clopidogrel resistance, changing to prasugrel therapy appears to confer significant benefit in terms of platelet reactivity and major adverse cardiovascular events [38, 160,164, 165]. Thus although sporadic reports of prasugrel resistance exist, there does not seem to be a clinical benefit to genetic testing in patients taking this drug [33].
Drug-Drug Interactions
As prasugrel increases bleeding risk, its use with other antiplatelet/anticoagulant medications, such as warfarin or long-term nonsteroidal anti-inflammatory drugs, may further exacerbate this risk. As CYP3A4/CYP3A5 accounts for most of prasugrel activation, inhibitors or inducers of this enzyme would be expected to impact bioavailability of the antiplatelet agent. However, for most drugs with a potential effect on this enzyme, there does not appear to be a clinically significant interaction. Prasugrel can be safely administered with common inhibitors of CYP3A4/CYP3A5, including statins, CCBs, or PPIs, without concern for impaired platelet inhibition [166]. Ketoconazole,a potent CYP3A4/CYP3A5 inhibitor that causes pharmacologic, although not clinical, clopidogrel resistance, does not appear to have a significant effect on prasugrel [87]. However, the protease inhibitor ritonavir, used commonly in patients with human immunodeficiency virus or hepatitis C virus infection, strongly inhibits both CYP3A and CYP2B6, leading to a significant reduction in prasugrel activation and bioavailability [167].Although the magnitude of the effect with ritonavir is similar to that with ketoconazole, clinical outcome data are lacking [168]. Thus caution is advised when ritonavir and prasugrel are used in patients with ACS [169].
Clinical Outcomes
The phase 2 Joint Utilization of Medications to Block Platelets Optimally (JUMBO)–Thrombolysis in Myocardial Infarction (TIMI)26 trial evaluated 904 patients undergoing PCI(excluding STEMI and shock). The patients were treated with aspirin at a dose of 325 mg and randomized to receive either clopidogrel (300 mg loading dose followed by 75 mg daily) or prasugrel (40 mg loading dose plus 7.5 mg daily, 60 mg loading dose plus 10 mg daily, or 60 mg loading dose plus 15 mg daily). The primary end point of this study, meant to evaluate safety, found no significant difference in non–coronary artery bypass graft (CABG) bleeding between those receiving prasugrel and those receiving clopidogrel at 30 days. Any bleeding, including TIMI major and minor bleeding, tended to be higher in the highdose prasugrel group, but the difference between the pooled prasugrel group and the clopidogrel group was not significant [170].
The Trial to Assess Improvement in Therapeutic Outcomes by Optimizing Platelet Inhibition with Prasugrel (TRITON)–TIMI-38 and its substudies evaluated 13,608 patients with moderate- to high-risk ACS, including NSTE-ACS [159, 171]and STEMI [172, 173]. In this population, they compared compare clopidogrel (300 mg loading dose and 75 mg daily) versus prasugrel (60 mg loading dose and 10 mg daily) in patients undergoing PCI. Notably, before randomization, the coronary anatomy had to be defined and deemed suitable for PCI. Exclusion criteria included increased risk of bleeding, anemia, thrombocytopenia, known intracranial abnormalities, or the use of thienopyridines in the previous 5 days. Prasugrel proved to be more efficacious, with the primary end point (composite of cardiovascular death, nonfatal MI, or nonfatal stroke) occurring in 12.1% of those receiving clopidogrel, compared with 9.9%of those receiving prasugrel. The prasugrel group also showed a significant reduction in death from cardiovascular causes, urgent target vessel revascularization, or rehospitalization for ischemia. The benefit was seen as early as day 3, and continued through the remainder of study period across all types of ACS [174]. These findings were consistent, regardless of stent type (drug-eluting stent vs.bare metal stent), and the rates of stent thrombosis were also lower with prasugrel (1.13 vs. 2.35%)across all groups [175]. The use of glycoprotein IIb/IIIa inhibitors did not diminish this benefit,as prasugrel significantly reduced the risk of MI,urgent revascularization, and stent thrombosis irrespective of glycoprotein IIb/IIIa inhibitor use[176]. Furthermore, patients with diabetes seemed to derive a greater benefit, with a greater reduction in ischemic events without an observed increase in TIMI major bleeding [177].
These impressive benefits, however, came with a significant increase in the risk of bleeding for patients taking prasugrel. The initial signal for increased bleeding came from the JUMBO-TIMI 26 trial [170], where there was a nonsignificant trend toward increased TIMI major and minor bleeding. In TRITON-TIMI-38, 2.4% of those taking prasugrel had non-CABG TIMI major bleeding, compared with 1.8% of those treated with clopidogrel, including fatal bleeding (0.4 vs. 0.1%).Additionally, in patients undergoing CABG, there was a significantly higher rate of bleeding in the prasugrel group (13.4 vs. 3.2%) [178]. Risk factors for bleeding included history of TIA/stroke,age more than 75 years, and body weight less than 60 kg, in addition to renal dysfunction and concomitant use of medications that increase the risk of bleeding [179]. Because of the increased bleeding risk, those with a previous TIA/stroke,age more than 75 years, and body weight less than 60 kg had no clinical benefit from prasugrel, especially because of the high morbidity and mortality of bleeding events in older patients [180].
In patients with ACS who were treated without intervention, there was no clear benefit from the use of prasugrel over clopidogrel. In the Targeted Platelet Inhibition to Clarify the Optimal Strategy to Medically Manage Acute Coronary Syndromes(TRILOGY-ACS) trial, prasugrel did not significantly reduce the frequency of a composite of death from cardiovascular causes, nonfatal MI, or nonfatal stroke in patients younger than 75 years, despite more potent platelet inhibition than clopidogrel[181, 182]. Building on this data, the Testing Platelet Reactivity in Patients Undergoing Elective Stent Placement on Clopidogrel to Guide Alternative Therapy with Prasugrel (TRIGGER-PCI) trial discovered that switching from clopidogrel to prasugrel in patients with high on-treatment platelet reactivity was not a clinically useful strategy in the era of modern drug-eluting stents, despite more potent platelet inhibition [183]. Furthermore, the A Comparison of Prasugrel at the Time of Percutaneous Coronary Intervention or as Pretreatment at the Time of Diagnosis in Patients with Non-ST Elevation Myocardial Infarction (ACCOAST) trial suggested that administration of prasugrel before PCI was not effective in reducing ischemic events and could be harmful because of the increased risk of bleeding compared with the administration of a loading dose at the time of PCI [184].
Thus the role of prasugrel in PAD or stable CAD is not well established, because of the lack of clinical efficacy and safety data in these conditions[185, 186]. On the basis of these findings, the current ACC/AHA guidelines offer only a class I recommendation for the use of prasugrel in patients presenting with ACS undergoing PCI (after the anatomy has been defined), adding a class IIb recommendation to choose prasugrel over clopidogrel for maintenance P2Y12inhibitor therapy [103].Prasugrel is contraindicated in patients with stroke,age more than 75 years, and body weight less than 60 kg. The role of prasugrel in stable CAD, PAD,and patients with atrial fibrillation undergoing PCI remains unclear.
Cyclopentyltriazolopyrimidines
Ticagrelor
Pharmacology
Ticagrelor is the first of a new class of drugs, known ascyclopentyltriazolopyrimidines, that also inhibit platelet aggregation by binding to the P2Y12inhibitor [187, 188]. However, there are key differences in terms of its metabolism and mechanism of effect,leading to a drug profile distinct from that of the thienopyridines. Ticagrelor, given with a 180 mg loading dose followed by a 90 mg maintenance dose twice daily, is not a prodrug, and therefore does not require metabolic activation to have a clinical effect [42, 189]. In bypassing the need for hepatic transformation, ticagrelor is able to quickly and consistently inhibit platelet aggregation [190].The ingested compound is absorbed in the small intestine, and, like clopidogrel, is subject to efflux via P-glycoprotein (encoded byABCB1) [191].After absorption, more than 99% of the parent compound binds to plasma proteins, with an estimated bioavailability of 36% [162, 192]. The initial antiplatelet effect occurs within 30 min, reaching the peak effect at approximately 2 h [193]. The mean plasma half-life of ticagrelor is 8 h, with elimination primarily via the gastrointestinal system [194].Although activation is not required for the platelet inhibitory effect of ticagrelor, it does undergo significant hepatic metabolism via CYP3A4/CYP3A5,leading to potential drug interactions [195]. This produces the active metabolite AR-C124910XX,which is at least as potent an antagonist of the P2Y12receptor as ticagrelor and is present in the circulation at approximately one-third of the concentration of the parent drug [192, 196]. The active metabolite reaches peak plasma concentration at 3 h, with a mean plasma half-life of 12 h after a loading dose[193]. Together, these compounds exhibit predictable linear pharmacokinetics, resulting in a rapid and potent clinical effect in a dose-dependent manner,achieving 80–90% platelet inhibition at peak concentration [188, 197]. Ticagrelor binds to the P2Y12receptor at a site distinct from the ADP-binding active site [198]. Rather than competitively inhibiting the ADP-P2Y12interaction, ticagrelor inhibits conformational change and G protein activation,rendering the receptor locked in the inactive state[4]. This inactive receptor is thus unable to promote the Giprotein–coupled cascade that culminates in platelet aggregation. No dose adjustments are needed on the basis of age or renal function [199,200]. Given the reversible nature of this drug, discontinuation of use of the drug results in a rapid offset, with platelet function returning to normal within 2–3 days [193].
Pleiotropic Effects of Ticagrelor
In addition to the antiplatelet effect of P2Y12inhibition, ticagrelor also inhibits adenosine reuptake,leading to coronary vasodilation and other potential unintended effects [201]. Ticagrelor is structurally similar to adenosine [202], and inhibits adenosine reuptake by erythrocytes via equilibrative nucleoside transporter 1 [203]. It is postulated that the increase in endogenous adenosine levels also contributes to ticagrelor’s antiplatelet activity [204].Furthermore, in animal models it was noted that ticagrelor, but not clopidogrel, limited infarct size by augmenting coronary blood flow through vasodilation [205, 206]. This finding was supported by evaluation of adenosine-mediated increases in coronary blood flow in healthy males receiving ticagrelor [204], thought to be due to upregulation of endothelial nitric oxide synthase and increased cAMP levels [206, 207]. Attempts have been made to implicate the increased endogenous adenosine with several of the potential adverse effects associated with ticagrelor, including dyspnea and bradycardia, although the role of this interaction is unclear. Specifically, dyspnea typically abates with continuation of therapy, and most of the bradyarrhythmias appear to be of sinoatrial origin,nocturnal, and typically do not require intervention [202]. There are also reports to suggest other off-target effects, including increased serum creatinine levels, improved endothelial function, and protection from the consequences of pulmonary infection, although debate remains regarding this issue [208].
Pharmacogenetics
Unlike clopidogrel, ticagrelor is not subject to significant modulation by the aforementioned common genetic variants that lead to clopidogrel resistance[191, 209]. In several large analyses ofCYP2C19andABCB1genetic polymorphisms did not have a significant impact on the pharmacologic or clinical profile of ticagrelor, with no significant effect on platelet inhibition or patient outcomes [210].Several single nucleotide polymorphisms affecting ticagrelor plasma levels were identified in genomewide association studies in the Platelet Inhibition and Patient Outcomes (PLATO) trial, although they were not associated with clinical outcomes[211, 212].
Drug-Drug Interactions
CYP3A4/CYP3A5 metabolism yields the ticagrelor active metabolite, which accounts for approximately 30% of the circulating drug, raising the concern that CYP3A4/CYP3A5 inhibitors/inducers may affect ticagrelor metabolism and its clinical effect.However, as ticagrelor is an active drug that does not require metabolic transformation to exert its antiplatelet effect, it is less susceptible to drug-drug interactions [213]. For example, coadministration of ticagrelor with atorvastatin or simvastatin does not appear to have any clinically relevant impact on the effect of any of these drugs, although an increase circulating simvastatin level was seen, warranting caution with doses greater than 40 mg [194, 214].Ticagrelor and its active metabolite are also substrates and inhibitors of P-glycoprotein, responsible for intestinal efflux of multiple drugs, including digoxin. Coadministration of ticagrelor and digoxin leads to increases (up to twofold) in digoxin concentration, warranting close monitoring of digoxin levels when ticagrelor is used [196]. Current recommendations discourage the use of strong CYP3A4 inhibitors (ketoconazole, voriconazole, clarithromycin, protease inhibitors) or inducers (rifampin,dexamethasone, phenytoin, carbamazepine) with ticagrelor, although moderate CYP3A4 inhibitors,such as diltiazem, can be safely coadministered[215, 216].
Clinical Outcomes
Coronary Artery Disease
The phase 2 Dose Confirmation Study Assessing Anti-platelet Effects of AZD6140 vs. Clopidogrel in Non-ST-segment Elevation Myocardial Infarction 2(DISPERSE-2) trial evaluated the safety and initial efficacy of ticagrelor (90 mg twice daily versus 180 mg twice daily) versus clopidogrel in 990 NSTE-ACS patients already receiving aspirin therapy. The trial showed no difference in major bleeding, but an increase in minor bleeding at the higher dose with a nonsignificant trend toward benefit with regard to the secondary end point, MI.Asymptomatic ventricular pauses longer than 2.5 s were more common with ticagrelor, particularly at 180 mg twice daily. Importantly, the trial demonstrated a decrease in major bleeding for patients undergoing CABG 1–5 days after stopping use of the drug, when compared to clopidogrel [217].More recent work demonstrates that the 180 mg loading dose led to more prompt and potent platelet inhibition [218]. The ONSET/OFFSET study evaluated the onset and offset of platelet inhibition with ticagrelor (180 mg loading dose) versus clopidogrel(600 mg loading dose) in 123 patients with stable CAD. A more rapid onset of platelet inhibition was seen with ticagrelor compared with clopidogrel as early as 30 min, with higher platelet inhibition in the maintenance therapy phase as well. Platelet inhibition was more consistent for patients receiving ticagrelor, with 90% achieving greater than 70%inhibition of platelet activity. Finally, a faster offset rate was observed after the last dose of ticagrelor than for clopidogrel [193].
The phase 3 PLATO trial and its substudies evaluated 18,624 patients with ACS (NSTEACS or STEMI) randomized to receive ticagrelor (180 mg loading dose followed by 90 mg twice daily) versus clopidogrel (300 mg loading dose followed by 75 mg daily), regardless of an invasive or conservative management strategy,representing an all-comers ACS population. In patients undergoing CABG, it was recommended that use of the study drug be withheld for 5 days in the clopidogrel group and for 24–72 h in the ticagrelor group. All patients received aspirin with a 325 mg loading dose (if thy were not already receiving the medication) followed by 75–100 mg daily. The primary efficacy end point was the composite of cardiovascular death, MI,and stroke; the primary safety end point included major bleeding defined according to study criteria. Ticagrelor was more effective than clopidogrel at reducing the primary outcome, driven by significant reductions in cardiovascular death and MI, with a significant 27% reduction in stent thrombosis. However, there was a statistically significant increase in non-CABG major bleeding and fatal intracranial bleeding, although net major (TIMI and by study criteria), life-threatening, and fatal bleeding was not statistically significant. Importantly, no specific subgroups had a higher rate of bleeding, including those with stroke [219]. PLATO substudies confirmed that this ischemic benefit was consistent in patients with STEMI, NSTE-ACS, planned invasive strategy, or noninvasive strategy, and in patients who underwent CABG, without significant increased bleeding [220–224]. Furthermore, the results held in patients with chronic kidney disease, in patients with diabetes, and in smokers, irrespective of age or angiographic outcome [225–229].Further analysis revealed a reduction in first and subsequent recurrent cardiovascular events, when compared with clopidogrel [230]. However,patients from North America experienced a reduced benefit with ticagrelor over clopidogrel,thought to be due to higher doses of aspirin in that group, although the cause of this interaction remains uncertain [231].
The subsequent Response to Ticagrelor in Clopidogrel Nonresponders and Responders and Effect of Switching Therapies (RESPOND)trial found that the antiplatelet effect of ticagrelor was consistent regardless of responsiveness to clopidogrel [232]. In 2015, the Prevention of Cardiovascular Events in Patients with Prior Heart Attack Using Ticagrelor Compared to Placebo on a Background of Aspirin (PEGASUS)-TIMI-54 trial demonstrated that patients with prior MI receiving aspirin therapy benefitted from the addition of ticagrelor, which led to a reduction in the risk of cardiovascular death, MI, or stroke when compared with placebo. In this trial, ticagrelor was associated with an increase in TIMI major bleeding, but not intracranial hemorrhage [233].The A 30 Day Study to Evaluate Efficacy and Safety of Pre-hospital vs. In-Hospital Initiation of Ticagrelor Therapy in STEMI Patients Planned for PCI (ATLANTIC) assessed the benefit of pretreatment with ticagrelor compared with placebo in STEMI patients, with co-primary end points of(1) ST-segment resolution on admission and (2)TIMI III flow before PCI. The trial demonstrated lack of superiority with ticagrelor, potentially due to the small difference in the time of administration (only 30 min) between the two groups.Importantly, more deaths occurred in the ticagrelor group (1.1 vs. 0.2%; P = 0.048) [234, 235].
As noted in the aforementioned studies, ticagrelor is associated with adverse events, including bleeding, dyspnea, and bradycardia. Dyspnea occurs in approximately 14% of patients taking ticagrelor,compared with 8% of patients taking clopidogrel.Although adenosine effects have been implicated as a cause, there remains debate on this issue.Recent analysis of the tolerability of this ticagrelor found that nearly one-third patients receiving 90 mg twice daily discontinued use of the medication, with approximately one-fifth citing adverse events, including bleeding (mostly nonmajor) and dyspnea [236].
Ischemic Stroke
In the initial PLATO trial, patients with prior stroke/TIA had higher rates of MI, death, recurrent stroke,and intracranial bleeding than patients without prior stroke/TIA, although efficacy and bleeding results of ticagrelor in these high-risk patients were consistent with the overall trial population [237]. The Acute Stroke or Transient Ischaemic Attack Treated with Aspirin or Ticagrelor and Patient Outcomes(SOCRATES) trial, looking at 13,199 patients with stroke or TIA, found that ticagrelor was not superior to aspirin in reducing the rate of stroke, MI, or death at 90 days [238]. However, a subgroup analysis revealed superiority of ticagrelor in patients with ipsilateral atherosclerotic stenosis, suggesting that the cause of stroke matters when one is choosing a drug for secondary prevention [239]. In this setting, the role of ticagrelor in stroke prevention remains uncertain.
Peripheral Artery Disease
Building on the potential role for clopidogrel in PAD, the Examining Use of Ticagrelor in PAD(EUCLID) trial studied 13,885 patients with symptomatic PAD randomized to receive monotherapy with ticagrelor (90 mg twice daily) or clopidogrel(75 mg once daily). The primary outcome (incidence of cardiovascular death, MI, or ischemic stroke) was no different in the two groups [240], with additional lack of efficacy in the cohort with a history of lower extremity revascularization [241]. Thus there is currently no clear role for ticagrelor in PAD.
With potential pleiotropic effects apart from platelet inhibition, ticagrelor confers a benefit over clopidogrel in terms of cardiovascular events and mortality in patients with ACS, regardless of invasive versus conservative management. On the basis of these findings, the current ACC/AHA guidelines recommend ticagrelor in patients with NSTE-ACS, independent of the management strategy, and in STEMI patients who have received a stent (class I), adding a class IIb recommendation to choose ticagrelor over clopidogrel for maintenance P2Y12inhibitor therapy [103]. On the basis of the PEGASUS-TIMI 54 trial, the guidelines also support continuation of P2Y12therapy beyond 12 months in prior MI patients who are not at high bleeding risk (class IIb). The role of ticagrelor in stroke, PAD, and patients with atrial fibrillation undergoing PCI remains unclear.
Cangrelor
Pharmacology
Although not an oral agent, the intravenous, reversible, rapid-acting P2Y12inhibitor cangrelor is an important component of modern antiplatelet therapy. Administered as an active compound that does not require activation, this cyclopentyltriazolopyrimidine medication exhibits a linear dose-dependent pharmacokinetic profile, resulting in platelet inhibition exceeding 90% [242]. Platelet inhibition typically begins within 2 min after administration of a bolus dose, with maximum inhibition achieved within 30 min [243]. With a half-life of 3–6 min, the drug undergoes deactivation to an inactive metabolite via dephosphorylation by a vascular endothelial endonucleotidase; this allows near complete platelet recovery within 60 min after discontinuation of the infusion [244].
Clinical Outcomes
Intended for use during PCI, cangrelor was investigated in a series of three related trials. The first of these, Cangrelor versus Standard Therapy to Achieve Optimal Management of Platelet Inhibition (CHAMPION) PLATFORM trial was a randomized, double-blind, placebo controlled trial that evaluated 5301 patients with unstable angina or NSTEMI undergoing PCI with or without stent implantation. Patients were randomized to receive either cangrelor (30 μg/kg bolus and 4 μg/kg per minute infusion) or placebo, followed by clopidogrel at a dose of 600 mg at the end of the infusion or procedure. From examination of the composite end point of death, MI, or ischemia-driven revascularization 48 h after PCI, cangrelor was not superior to placebo. However, the prespecified secondary end points of stent thrombosis and death were lower in the cangrelor group, with a signal for more major bleeding [245]. At the same time, the CHAMPION PCI trial compared cangrelor with 600 mg of orally administered clopidogrel given before PCI in 8722 patients with stable angina, unstable angina,NSTEMI, or STEMI. Again, cangrelor was not superior to 600 mg of clopidogrel when the composite primary end point of death from any cause,MI, or ischemia-driven revascularization at 48 h was examined [246].
Subsequently, the 2013 CHAMPION PHOENIX trial randomized 11,145 patients with stable angina,unstable angina, NSTEMI, or STEMI undergoing urgent or elective PCI to receive cangrelor or clopidogrel. In this trial, the primary outcome was a composite of all-cause death, MI, ischemiadriven revascularization, or, unlike the prior trials,stent thrombosis. In this setting, cangrelor significantly reduced the risk of the primary outcome without increasing in the risk of severe bleeding[247]. A prespecified pooled analysis of these three CHAMPION studies suggested that cangrelor was better than the control clopidogrel in decreasing post-PCI thrombotic complications [248].
On the basis of the findings of the CHAMPION PHOENIX trial, cangrelor gained Food and Drug Admiration approval for use during PCI. Notably,cangrelor treatment was associated with a higher risk of dyspnea, as with ticagrelor. At this point it remains unclear how cangrelor compares with ticagrelor and prasugrel.
New and Upcoming Data
The recent single-center observational CHANGEDAPT study (clopidogrel or ticagrelor in ACS patients treated with newer-generation drug-eluting stents) noted a higher rate of major bleeding in ticagrelor-treated ACS patients compared with clopidogrel-treated ACS patients (2.7 vs. 1.2%;adjusted hazard ratio 2.75; 95% confidence interval 1.34–5.61), without significant ischemic benefit for the newer-generation drug [249]. This furthers the findings of the TOPIC randomized study (benefit of switching DAPT after acute coronary syndrome), which found that a strategy of switching from prasugrel or ticagrelor to clopidogrel 1 month after PCI for ACS was associated with fewer bleeding events, without a significant change in ischemic outcomes [250]. Furthermore, the optimal duration of DAPT is undergoing more nuanced exploration.In ACS, the 6 versus 12 Months of Dual Antiplatelet Therapy after Drug-Eluting Stent Implantation in ST-Elevation Myocardial Infarction (DAPT STEMI) trial showed that among patients who remain event-free at 6 months following PCI with a zotarolimus-eluting stent for STEMI, cessation of DAPT at 6 months was noninferior to continuation to 12 months [251]. Many of these new data are focused on the role of newer-generation drugeluting stents, with smaller stent struts and a lower risk of stent thrombosis. In non-ACS PCI, a series of trials are evaluating 1–3 months of therapy with a P2Y12inhibitor with newer-generation stents[252, 253]. The findings of these studies may have significant impact on the use of DAPT in patients who do not present with ACS. Although not practice-changing, these studies suggest that the role of these newer antiplatelet agents in ACS will need to be further explored in randomized controlled trials, particularly with the advent of new stent technology.
Conclusion
Of the P2Y12inhibitors, clopidogrel is commonly prescribed in the United States, with an established track record of safety and efficacy in various conditions, including stable CAD, ACS, stroke, PAD, and atrial fibrillation as combined therapy. As a generic medication, its relative affordability makes it an appealing choice for many patients and providers.However, it is limited by common polymorphisms that promote high on-treatment platelet reactivity and thus a higher risk of recurrent ischemic events.Both ticagrelor and prasugrel are superior to clopidogrel in terms of ischemic outcomes in patients with ACS who undergo PCI. Preliminary evidence from a head-to-head comparison of these newer agents suggests that one is neither more effective nor safer than the other in this population [254,255]. Ticagrelor, however, is limited by cost and adverse effects, such as dyspnea, leading to medication use discontinuation. Prasugrel has a limited scope of indications and several contraindications to its use due to a higher risk of bleeding. In the United States, economic analysis of patients from the PLATO trial suggests that for PLATO-eligible ACS patients, an aspirin plus ticagrelor regimen increases life expectancy at an incremental cost well within accepted benchmarks of good value for money [256], although further analysis is still needed [257, 258]. Although ticagrelor and prasugrel are the preferred options in patients with ACS undergoing PCI, clopidogrel remains a cost-effective antiplatelet agent with the widest range of potential applications, suggesting that for many of our patients it remains an excellent first choice.
杂志排行

Cardiovascular Innovations and Applications的其它文章
- Speckle Tracking Echocardiography ldentifies lmpaired Longitudinal Strain as a Common Deficit in Various Cardiac Diseases
- Current Status of Coronary Atherectomy
- The Use of Direct Oral Anticoagulants for Prevention of Stroke and Systemic Embolic Events in East Asian Patients with Nonvalvular Atrial Fibrillation
- Bioresorbable Vascular Scaffold in the Midportion of the Left Anterior Descending Artery for Cardiac Allograft Vasculopathy in a Cardiac Transplant Patient
- The Contemporary Role of Femoral Artery Access
- Cardiovascular Innovations and Applications