Disprot无序蛋白数据库分析与统计
2018-09-17李盘靖
张 欢,李盘靖,王 彤
(山东理工大学 计算机科学与技术学院,山东 淄博 255049)
蛋白质在生物世界中扮演了各种各样的角色.传统思想认为,氨基酸序列决定蛋白质唯一的三维结构,三维结构则决定了蛋白质的生物学功能[1]形成了蛋白质科学的经典研究范式“序列-结构-功能”.20世纪90年代初,随着实验技术的发展,人们发现有些蛋白质或蛋白质序列中的一部分在天然状态下并不具有一个确定的三维结构,但依然具有正常的生物学活性.后来进一步研究发现这类蛋白质越来越多,并逐渐形成了一类与传统蛋白质范式不同的新的蛋白质类型,称为天然无序蛋白[2-3](intrinsically disordered proteins,简称为IDPs).根据无序蛋白中所含无序结构的多少,可以将无序蛋白分为两大类:完全无序蛋白(全序列无序)和部分无序蛋白(局部超过30~40个残基的区域无序);部分无序蛋白由结构域(structured domains)和无序区域组成(disordered regions)[4].无序蛋白中的无序结构与蛋白质功能之间关系密切[5],无序蛋白在诸如转录、翻译、调控细胞信号转导、蛋白质磷酸化及小分子存储等过程中发挥着重要的作用[2].另一方面,无序蛋白常与多种疾病联系在一起.无序蛋白的无序特性使得它可以与多种伴侣分子结合从而在分子网络中达到传递信号或是调节的作用,人类的许多疾病例如癌症、心血管疾病、神经性衰弱等不仅与相关功能性蛋白的误折叠有关信号之间的误传导、误表达有关[6].因此判定蛋白质的无序区成为蛋白质科学中的一个热点问题.Romero等在1997年首次对蛋白质无序区进行预测,他们预测的准确性达到70%[7].此后,无序蛋白质的预测方法得到了迅速发展,目前应用于无序蛋白质序列预测的方法已经超过50种,这些预测方法的准确性普遍达到85%以上.随着2012年深度学习方法在图像分类预测上成功的应用,近年来关于无序蛋白的研究又出现了新的热度,从2006年hinton重新提出深度模型后[8],无序蛋白的研究论文数快速增长.本研究基于序列分析的方法,以Disprot数据库中的无序蛋白序列为研究对象,通过CD-HIT去冗余程序处理后建立数据集(无序区和有序区),然后将得到的数据集通过Python语言进行统计分析,分别提取出无序区和有序区中的残基进行统计从而分析其偏好性.
1 Disprot数据库简介与使用方法
Disprot数据库从已有文献中搜集经过实验验证的无序蛋白数据,每一条蛋白质序列都标注了无序片段的起始位置、判定方法、来源文献,该无序片段行驶的生物学功能也进行了标注.网站中提供了csv、json两种格式的数据格式文件.在进行后期的无序片段标注时,要组合蛋白质与无序片段数据进行下载.然后通过编程工具python进行脚本标注无序片段,分别建立数据集.数据库现包含803条无序蛋白数据,所属分类如图1所示.

图1 无序蛋白所属的4个主要分类Fig.1 4 major superkingdoms of intrinsically disordered proteins
从图1可以看出,无序蛋白主要分布在真核生物、细菌当中,少部分存在于病毒与古生菌中.
2 数据集的创建与序列分析方法
2.1 数据集
本研究中固有无序蛋白序列数据取自固有无序蛋白数据库[9].当前版本中含有803条IDPs蛋白质链,2167个无序区.由于蛋白质数据库中含有大量的冗余序列,不利于数据的统计分析,我们利用去冗余程序CD-HIT[10]对数据进行处理,将相似度阈值设为30%.结果显示,去冗余前,该数据库中共有803条序列;去冗余后,减少到708条序列.
2.2 序列分析方法
2.2.1 氨基酸分别在无序区与有序区中的分布
根据数据库中的708条序列分析,氨基酸残基总数为363575,其中有序区的氨基酸残基总数为280852(占77.3%),无序区的氨基酸残基总数为82723(占22.7%).将无序区残基与有序区残基的分布绘制在图2中.
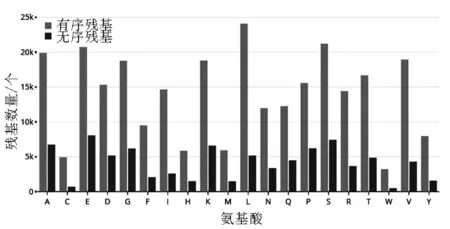
图2 20种氨基酸在无序与有序序列中的分布Fig.2 The distribution of 20 kinds of amimo acids in ordered and disordered region
由图2可以看出20种氨基酸在无序区与有序区中的分布具有相似性,其相似性通过KL-散度定量得到.KL-散度的定义公式为
(1)
KL-散度值越接近0,说明两种分布越相似.式中P代表无序区中氨基酸的分布;Q代表有序区中氨基酸的分布.
2.2.2 氨基酸的无序倾向性
定义氨基酸形成无序区的倾向性公式为
DP(x)=dp(x)-op(x)
(2)
式中:DP(x)(disorder propensity)表示氨基酸x在无序区出现的倾向性;dp(x)表示氨基酸x在无序区中出现的几率;op(x)表示氨基酸x在有序区中出现的几率.
根据DP(x)值的大小,可以判断氨基酸形成无序区的偏好性.DP(x)>0,该氨基酸具有形成蛋白质无序区的倾向性;DP(x)<0,该氨基酸具有形成蛋白质有序区的倾向[11].
2.2.3 二元组氨基酸对统计
在上述单个氨基酸统计的基础上,增加了二元组氨基酸对的统计.根据Disport数据库对无序序列的标注,抽取出无序序列,然后编程实现对每一条无序序列二元组的统计.
3 结果及讨论
3.1 氨基酸在无序区与有序区中的分布
由图2得到20种氨基酸的分布图可以看出:有序区和无序区中Ala、Asp、Glu、Gly、Lys、Leu、Pro、Ser、Thr、Val都有很强的倾向性.通过式(1)与图2的数据得到无序区和有序区氨基酸分布的KL-散度值为0.031,说明无序区和有序区中氨基酸的分布具有相似性,可以推断随着数据集的不断扩充,这种相似性会越来越高.20种氨基酸在无序区和有序区具有分布相似性,还需要接下来DP值的分析来判断氨基酸的无序倾向性.
3.2 氨基酸形成无序区的倾向性分析
根据式(2)计算了20种氨基酸的DP值,如图3所示.
根据DP值的大小,可以看出氨基酸Ala、Asp、Glu、Gly、Lys、Pro、Gln、Ser具有形成无序区的倾向.在第一步分析中氨基酸Leu、Thr、Val虽然在无序区和有序区中都具有倾向性,但在由DP值得到的分析中,Leu、Thr、Val不易于形成无序区;氨基酸Gln虽然在无序区和有序区中都不具有倾向性,但在DP值分析中却易于形成无序区.在表1中氨基酸各性质的描述中可以看到,具有无序倾向的8种氨基酸用黑体标出.8种氨基酸都属于非芳香或脂肪族氨基酸,亲水性氨基酸与疏水性氨基酸之比为3∶2,大部分疏水性氨基酸都不具有无序倾向性.在8种无序倾向氨基酸中极性与非极性氨基酸之别为5∶3.

图3 20种氨基酸的无序倾向性DP值Fig.3 The DP(disorder propensity) values of 20 kinds of amino acids
表1 氨基酸的性质
Tab. 1 Attributes of amino acids

缩写全名中文译名支链极性芳香或脂肪族GlyGlycine甘氨酸亲水性--AlaAlanine丙氨酸疏水性--ValValine缬氨酸疏水性-脂肪性LeuLeucine亮氨酸疏水性-脂肪性IleIsoleucine异亮氨酸疏水性-脂肪性PhePhenylalanine苯丙氨酸疏水性-芳香性TrpTryptophan色氨酸疏水性-芳香性TyrTyrosine酪氨酸亲水性X芳香性AspAspartate天冬氨酸酸性X-HisHistidine组氨酸碱性X芳香性AsnAsparagine天冬酰胺亲水性X-GluGlutamate谷氨酸酸性X-LysLysine赖氨酸碱性X-GlnGlutamine谷氨酰胺亲水性X-MetMethionine甲硫氨酸疏水性--ArgArginine精氨酸碱性X-SerSerine丝氨酸亲水性X-ThrThreonine苏氨酸亲水性X-CysCysteine半胱氨酸亲水性--ProProline脯氨酸疏水性--
3.3 二元组氨基酸对的分布
根据以下二元组抽取代码为基础,统计得到402对二元组氨基酸对.
# 从Disprot数据库中抽取无序序列到disorder_seq
# 二元组氨基酸对统计结果保存在 bi_key 中
bi_key = dict()
for seq in disorder_seq:
keys = [ seq[i:i+2] for i in range(0, len(seq)-1) ]
for key in keys:
if key not in bi_key:
bi_key[key] = 1
else:
bi_key[key] = bi_key[key] + 1
统计得到使用最频繁的20种氨基酸对如图4所示.由之前DP值的分析中Asp、Glu、Ser易于形成无序区,在二元组统计中,由这3种氨基酸组成的二元组也最频繁使用.在无序蛋白无序序列中偏向于使用简单重复的氨基酸对Ala、Glu、Ser.

图4 使用最频繁的20种氨基酸对Fig.4 The most used 20 amino acids tuples
4 结束语
以蛋白质序列信息为基础,通过分析20种氨基酸在无序区与有序区中的分布和氨基酸的无序倾向性,为下一步开发无序序列预测算法[12]提供了特征准备.本文只在序列的角度总体上统计了各氨基酸的分布,但是一个无序位点的形成不仅与它本身的特征有关,还与它的上下文位点存在关联.本文只讨论了二元组的情况下氨基酸对的无序倾向性.在下一步工作中, 多位点之间的联合特征提取是一个重要的研究方向.
