上海地区汉族健康成年人唾液微生物菌群结构分析
2018-09-10蔡丽婷邵方洋郑赛巍汪丙杰
史 聪, 蔡丽婷, 邵方洋, 郑赛巍, 汪丙杰, 何 园
(1. 同济大学口腔医学院,上海 200072; 2. 同济大学附属口腔医院黏膜病学教研室-上海牙修复与再生工程实验中心,上海 200072; 3. 同济大学生命科学与技术学院生物信息学科,上海 200092)
口腔疾病的发生发展均与微生物群落结构失调有关。深入了解健康状态下的口腔微生物群落特征,有助于进一步认识疾病状态下的微生物群落特征,利于疾病的早期诊断及疗效监测。口腔内的微生物群落有细菌、病毒、真菌、支原体、古细菌和原生生物等[1],其中细菌的群落结构非常复杂,目前检测到的大约有1000种。近年来,随着高通量测序技术及宏基因组学发展迅速,使得人们对微生物的认识有了全新的视角。本研究拟采用宏基因组学技术,分析健康人的唾液微生物群落结构,为口腔疾病的预防及诊治提供参考。
1 资料与方法
1.1 一般资料
37名符合纳入标准的受检者均为上海地区的常住汉族居民,其中男性20名,女性17名,见表1。所有受检者均由两名经培训的口腔专业医生依据1997年WHO口腔健康调查基本方法进行临床检查。受检者的纳入标准为: DFT(decay-filled index, DFT)=0;CPI(community periodontal index, CPI)=0。排除标准为: 患有其他口腔疾病(如口腔黏膜病、唾液腺疾病等);口腔内有修复体;少于28颗牙齿;患有全身系统性疾病及肿瘤;近1个月内使用抗生素或抗真菌药物;吸烟;女性处于月经期、妊娠期或哺乳期。所有受检者均在取样前签署了知情同意书,并且该研究得到了同济大学医学伦理道德委员会的批准。

表1 37名受检者的样本信息Tab.1 Characteristics of 37 individuals in this study
1.2 样本采集
所有受检者均在样本收集前完成必要的口腔检查及样本信息登记表。取样前嘱受检者禁食2h。受检者清水漱口1min,采集1.5mL非刺激性唾液,封口后立即置于-20℃冰箱中保存,并且随后转入-80℃冰箱中长期保存。
1.3 唾液样本总DNA提取,PCR扩增及测序
口腔唾液总DNA采用QIAamp DNA micro试剂盒提取(凯杰企业管理有限公司),提取方法参照试剂盒使用说明,随后用分光光度计检测提取物的质量和浓度。随后采用TRANS公司的TransStartFastpfu DNA Polymerase,20μL反应体系对V3~V4可变区进行PCR扩增(北京全式金生物技术有限公司),并且应用2%琼脂糖凝胶电泳检测纯化后PCR产物。16 S rRNA的V3~V4可变区通用引物设计如下。上游引物为5′-CCTAYG-GGRBGCASCAG-3′;下游引物为5′-GGACTACNN-GGGTWTCTAAT-3′。最后,由Illumina HiSeq 2500测序平台对扩增产物进行测序(上海凌恩生物科技有限公司,中国)。
1.4 数据处理
采用Trimmomatic和QIIME(version 1.8)对序列进行处理和分析。本研究过滤掉了序列长度小于200bp的序列,并且采用USEARCH方法鉴别并过滤掉嵌合体序列。按照97%的相似度划分操作分类单元(operational taxonomic units, OUTs),并与Greengenes数据库进行比对,通过RDP分类器对OUT进行逐级分类。运用QIIME计算稀释曲线和多样性指数,并应用Unweighted UniFrac方法计算样本差异矩阵距离,然后进行主成分分析(PCoA)。定义一级核心菌属,标准为: 在所有样本中均有分布,且在样本中相对丰度均值≥2%的菌属[2]。并且应用iTOL对主要菌属(平均相对丰度>0.1%)构建唾液菌丛系统发育树。P<0.05为差异有统计学意义。
2 结 果
2.1 样本信息及基因组DNA序列分析
本研究通过Illumina HiSeq测序平台测序共获得1329271条优质序列,每个样本平均为(35926.24±4718.069)条。这些序列可归为110个属,12个门,90.78%的序列可以分类到72个已知属,测序质量较高,可以用于后续数据分析。根据observed_species数据,做出每个样本的稀释曲线,曲线均趋于平坦,说明本次实验测序深度达到要求,见图1。
2.2 唾液微生物结构
健康青年人的口腔唾液菌群科分类到12门。其中5个高丰度菌门(相对丰度>5%)占总序列数的97.54%,分别为: 厚壁菌门(Firmicutes,42.21%)、变形菌门(Proteobacteria,27.78%)、拟杆菌门(Bacter-oidetes,14.34%)、放线菌门(Actinobacteria,7.96%)、梭杆菌门(Fusobacteria,5.25%)。除了5个高丰度菌门以外,还有3个菌门在所有样本中均有分布,分别为TM7、螺旋体门(Spirochaetes)、SR1。健康青年人口腔微生物分布相对集中,以上8个核心菌门在总序列中所占比例为99.69%,见图2。
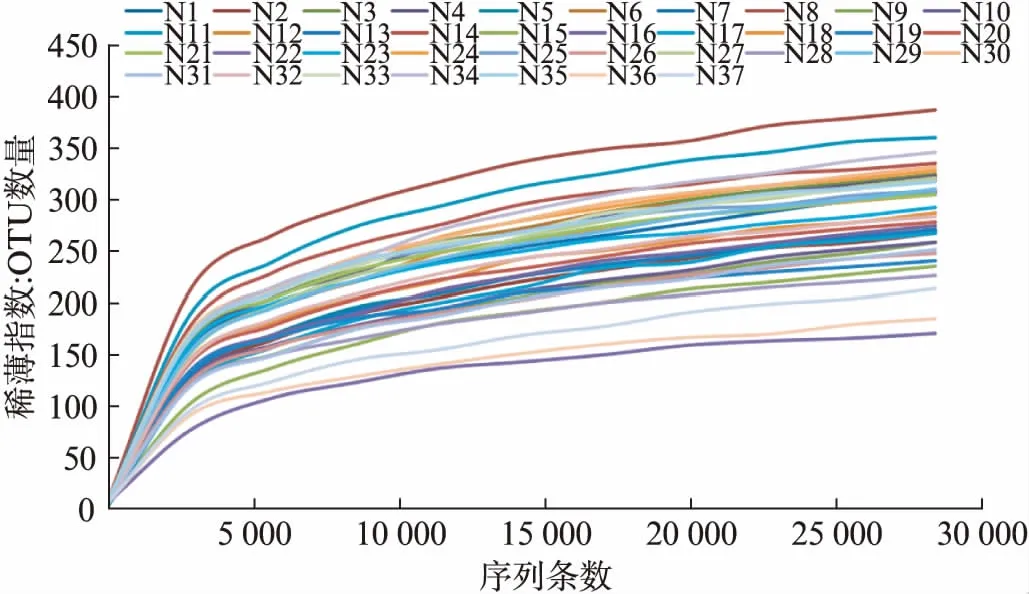
图1 稀释曲线Fig.1 Rarefaction curves of the oral microbiota稀释曲线可以反映物种多样性,由图可知observed-species数量随着序列条数增加而增加,最后达到平台期,说明本研究深度达到要求
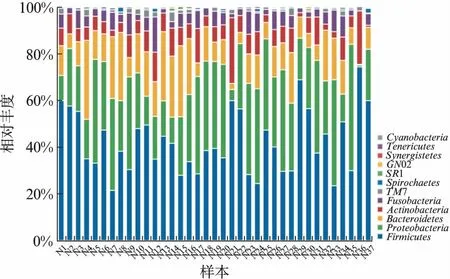
图2 唾液微生物门在样本中的分布Fig.2 Phylum-level relative abundance健康组菌群可分类到12个门,97.54%的序列属于5个高丰度菌门,占序列比例为99.69%的8个核心菌门在所有样本中均有分布,说明健康青年人口腔微生物分布相对集中
健康青年人的口腔唾液微生物在属水平的分布也相对集中,主要微生物(相对丰度>10%)为链球菌属(25.90%)和奈瑟菌属(13.05%)。根据对核心菌的定义共得到12个核心菌属,占微生物总序列的79.71%。分别为: 链球菌属(Streptococcus)、奈瑟菌属(Neisseria)、嗜血菌属(Haemophilus,7.62%)、普氏菌属(Prevotella,6.58%)、罗氏菌属(Rothia,4.53%)、卟啉单胞菌属(Porphyromonas,4.02%)、Gemellaceaegen.、韦荣菌属(Veillonella,3.41%)、颗粒链球菌属(Granulicatella,3.09%)、梭杆菌属(Fusobacterium,2.74%)、放线菌属(Actinomyces,2.52%)、纤毛菌属(Leptotrichia,2.46%),见图3。为了更清楚地表达唾液的微生物群落结构,进一步对主要菌属(平均相对丰度>0.1%)构建唾液菌丛系统发育树,见图4。

图3 样本中核心微生物Fig.3 The relative abundance of core microbiome in samples在属水平,健康人的唾液微生物菌群结构分布也比较集中,相对丰度>10%的有链球菌属和奈瑟菌属
2.3 唾液微生物多样性分析
各样本的序列覆盖度均在99.7%以上,说明样本的测序量达到要求,基本覆盖所有微生物。且男性与女性比较中,Shannon多样性指数差异无统计学意义(P>0.05),说明男性与女性之间α多样性没有差异,见图5A。各样本的主成分分析比较集中,且男性与女性在各自象限有交叉,说明男性与女性之间菌群结构相似,见图5B。
3 讨 论
唾液菌群是机体内环境的重要组成部分,其结构在健康状态下保持相对稳定及与宿主之间的稳态是维持口腔健康必不可少的条件。然而,当这种平衡被某些原因打破后,菌群结构失调将引发疾病的发生。目前发现与微生物有关的疾病包括龋病[3]、牙周炎[4]等。还有些口腔微生物甚至影响全身健康,如有学者发现链球菌(Streptococci)与糖尿病有关[5],糖尿病严重影响患者的生活质量[6]。只有对健康状态下的口腔唾液菌群菌群结构有明确的认识,才能更好的发现疾病状态下微生物菌落结构的改变及这种改变的影响。

图4 健康成年口腔唾液菌丛属水平系统发育树Fig.4 Circular phylogenetic tree of saliva from healthy adults at genus level选取属水平菌平均相对丰度>0.1%者,共有47个属;左侧图例为科水平,右侧图中内圈不同颜色代表不同的科水平菌,同一科的属用相同颜色表示,外带显示相对丰度
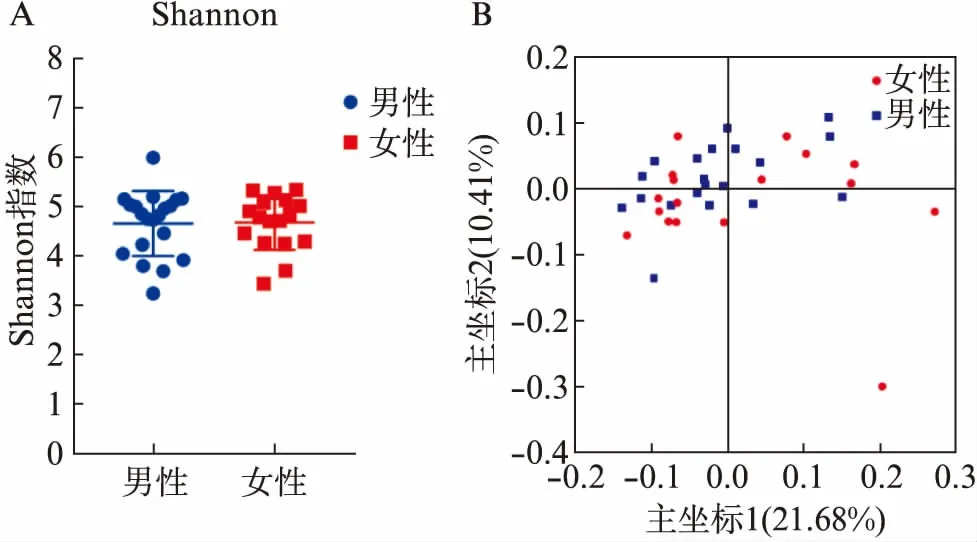
图5 健康人唾液菌群多样性分析Fig.5 Analysis of the diversity of salivary microbiota in male and female subjectsA: 健康的男性与女性唾液样本的Shannon多样性指数分析,可见男性与女性之间菌群多样性无差异;B: 健康的男性和女性两组唾液样本的主坐标分析,可见男性与女性之间菌群结构没有差异
正常情况下,整个口腔,包括牙齿与黏膜均浸润在唾液中,可以说唾液构成了口腔的外环境。研究[7]表明,唾液微生物组受唾液采集方法、储存及DNA提取的影响很小。因此,唾液是研究口腔微生物群落的重要生物样本[8]。
近年来,高通量测序技术以及宏基因组学的发展使得人们对人类微生物组研究领域发生了革命性的进展,尤其是对肠道微生物的研究[9]。相对于以往基于细菌培养的方法而言,高通量测序技术使得许多不能等培养的微生物得以被检测到。与人类口腔微生物鉴别芯片(human oral microbe identification microarray, HOMIM)相比,高通量测序技术可以检测到大量的低丰度物种[10],如SR1、Tenericutesspp.和Synergistetesspp.。本研究测序的序列覆盖度在99.7%以上,平均每个样本为(35926.24±4718.069)条序列,并且稀释曲线达到平台期,比以往的研究更深入地检测了口腔微生物菌群。
Segata等[11]和其他HMP(Human Microbiome Project, HMP)成员在一项由美国国立卫生研究院设立的人类微生物组计划中,通过对V1~V3和V3~V5 16 S rRNA高变区进行测序,探索了超过200个个体的18个身体部位(包括口腔唾液)。这一大型的队列研究项目明确了超过95%的口腔唾液微生物是由5个主要门组成: 厚壁菌门、变形菌门、拟杆菌门、放线菌门、梭杆菌门。在本研究中,健康人的唾液微生物组成与HMP的研究结果一致,说明本次测序结果是可靠的。
核心微生物组是指所有或者绝大多数样本共有的微生物[12]。核心微生物代表了口腔微生物的组成及菌群结构,一个健康的口腔微生物群应该由一个健康的“核心微生物”主导。这些含量丰富的核心微生物将保持健康生态系统所必需的功能稳定性和体内平衡。目前来看,在属水平定义核心微生物组比在门水平定义更加合理。因此,本研究定义了一级核心菌属,标准为: 在所有样本中均有分布,且在样本中相对丰度均值≥2%的菌属[2]。根据对核心菌的定义共得到12个核心菌属: 链球菌属、奈瑟菌属、嗜血菌属、普氏菌属、罗氏菌属、卟啉单胞菌属、Gemellaceaegen.、韦荣菌属、颗粒链球菌属、梭杆菌属、放线菌属、纤毛菌属。
链球菌属、奈瑟菌属、放线菌属、韦荣菌属、颗粒链球菌属是与健康息息相关的,这个观点在第十二届欧洲牙周病研讨会中被达成共识[13]。格登链球菌(S.gordonii)是口腔生物膜群落的重要组成成分,作为口腔共生菌,可以促进生物膜的稳态的形成[14],并且可以增加上皮屏障功能,有利于维持健康组织上皮的完整性。血链球菌(S.sanguis)也是常见的口腔共生菌,其产生的过氧化氢和血链素被认为可以拮抗牙周致病菌[15]。韦荣菌属也是参与口腔生物膜形成的早期定植菌,可与链球菌属共聚,促进格登链球菌生物膜的形成[16]。放线菌属可与血链球菌共聚,保护其免受氧化损伤[17]。并且韦荣菌属、奈瑟菌属、放线菌属在许多研究中均被证实为健康人口腔中的优势菌[4,18]。
本研究在核心微生物中也发现大量常见的与人类疾病相关的微生物。其中,卟啉单胞菌属属于“红色复合体”,与牙周炎的发生发展密切相关[19]。还有证据表明,牙龈卟啉单胞菌可能是包括口腔癌、肠癌和胰腺癌在内的消化道肿瘤发展的驱动因素,口腔鳞状细胞癌(OSCC)也与其高度相关[10]。F.nucleatum是梭杆菌属中调查最广泛的物种,研究[20]表明,F.nucleatum可以在人类牙齿上形成典型的牙菌斑,与牙周病有关,并可引起人类头颈部、胸部、肺部、肝脏和腹部的侵袭性感染。罗氏菌是在龋洞中发现的,且在磨牙龋洞中的检测率很高,表明其与龋病相关[21]。纤毛菌属可存在于人体口腔及生殖道黏膜,进入血液循环,可引发严重的脓毒血症。这些微生物在健康状态下不具有致病性,但是在机体免疫功能降低或口腔菌群结构失调时引发疾病影响宿主的健康。因此,健康人口腔微生物的研究,可促进对口腔微环境的了解,在疾病预防和早期诊断及治疗中有重要意义。
