基于高通量测序分析河北省中南部地区耕地土壤细菌多样性
2018-09-10宋水山黄亚丽贾振华黄媛媛
张 翔,宋水山,黄亚丽,贾振华,黄媛媛,宋 聪
(1.河北省科学院 生物研究所,河北 石家庄 050081;2.河北科技大学 生物科学与工程学院,河北 石家庄 050000;3.河北省主要农作物病害微生物控制工程技术研究中心,河北 石家庄 050081)
河北省地处华北平原,土地总面积188 544.71 km2,其中,耕地面积有6 537.74 km2[1]。河北省平原地区主要以温室大棚和大田2种培养方式进行种植。温室大棚是一种不受外界环境影响、能够人工调节局部环境条件,来达到农作物生长最合适的生态环境的种植方式[2]。温室大棚可以延长蔬菜的生产时间,增加土地利用率,提高产量。但是,温室大棚因为其封闭性、局限性,使大棚的内部长期处于高温、高湿的环境中[3]。这就使得大棚中的土壤会随着耕种年限的延长,出现土壤酸化、盐渍化、土壤微生物结构失衡、易染病、土壤养分失衡、重金属和农药化肥积累等问题[4-7],会严重影响农作物的产量。
在温室大棚生态环境中,土壤微生物的群落组成能够较为准确地反映大棚土壤的状态。土壤微生物种类繁多、结构复杂,而目前可培养的微生物仅占到了1%~10%,并不能反映土壤微生物的群落结构[8]。最新的MiSeq高通量测序平台可以对土壤中所有微生物的16S rDNA、18S rDNA及ITS等序列进行测序,并对土壤中的优势物种、稀有物种和未知物种进行检测,分析每个物种的相对丰度及微生物群落的组成[9-12]。
目前,对温室大棚与露天大田的土壤微生物群落结构的对比很少有报道。本研究利用高通量测序技术对河北省中南部地区的温室大棚与露天大田的土壤微生物群落结构进行对比分析,旨在进一步了解2种种植方式对土壤微生物群落结构的影响,为改善温室大棚土壤环境,提高抗病性,提高农作物产量提供依据。
1 材料和方法
1.1 样品采集
试验所用土壤样品均采集于河北省平原地区的温室大棚土壤及其周边的大田土壤。土壤样品的收集方法采用5点采样法,采集深度0~10 cm。样品取好后放入冰盒中带回实验室,并放入-80 ℃冰箱中储存备用[13]。
各个土壤样品的采集信息见表1。
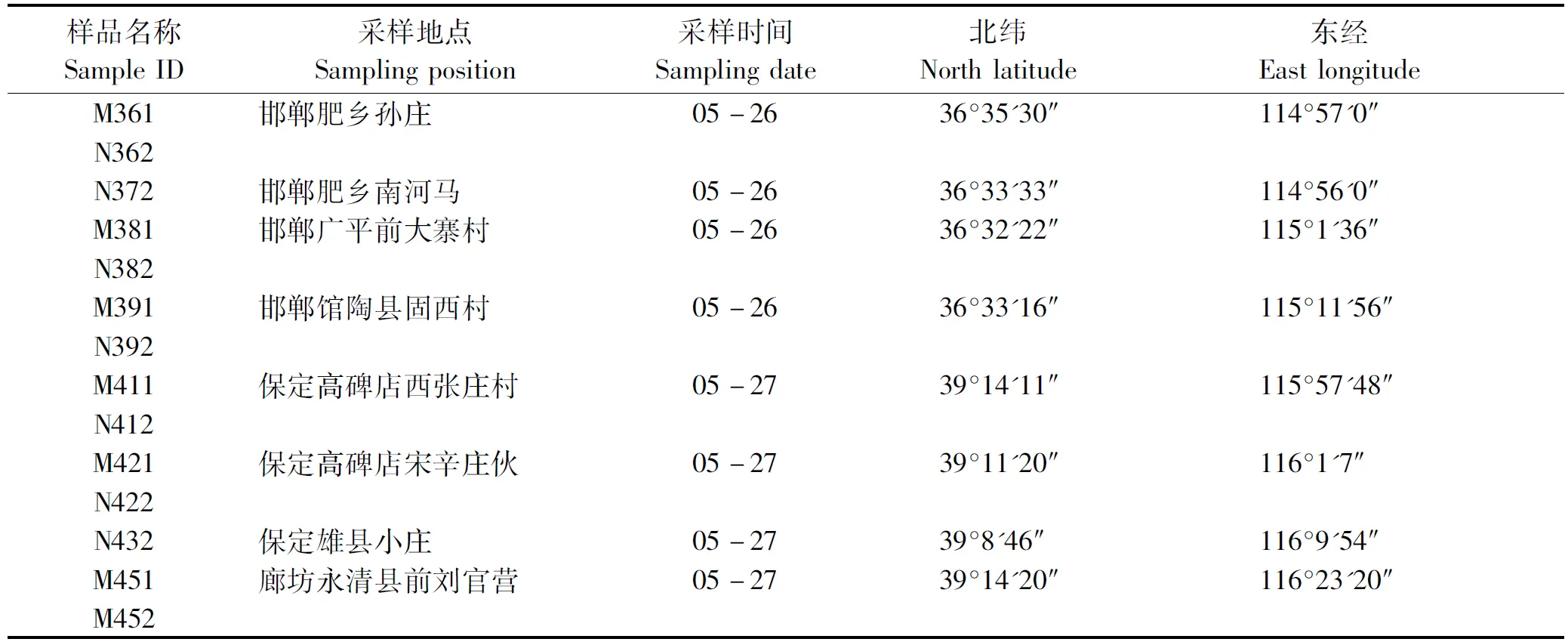
表1 土壤样品采集信息(2016年)Tab.1 Soil samples collection information table
1.2 土壤中细菌总DNA的提取及PCR扩增
从带回的土壤样品中取0.25 g,利用Mobio公司的Powersoil土壤DNA提取试剂盒进行土壤总DNA的提取。DNA提取好后,用通用引物338F(5′-ACTCCTACGGGAGGCAGC-3′)和806R(5′-GGACTA CHVGGGTWTCTAAT-3′)对土壤中细菌16S rDNA的V3-V4区域进行PCR扩增。
1.3 MiSeq上机测序
将纯化好PCR产物送去北京奥维森基因科技有限公司,利用 Illumina Miseq 高通量测序平台进行测序和分析。
1.4 数据分析
首先,利用Trimmomatic、FLASH、Pear、usearch软件对数据进行拼接、质控和去除嵌合体。然后,对拼接好的序列进行进一步的去除嵌合体和段序列得到优质序列。接下来,利用Qiime、uclust、usearch软件按照97%相似性将全部优质序列进行聚类,得到代表序列和OTU表。利用mothur等软件进行稀释性曲线的绘制,利用Qiime等软件计算Chao1值、observed_species指数、goods_coverage指数和Shannon指数。为了得到每个OTU的物种信息,采用RDP Classifier算法对每个OTU代表序列进行比对分析,并在各个水平注释群落物种信息。利用R语言绘制物种组成分析和PCA分析的图表[14]。
2 结果与分析
2.1 测序结果分析
采集的14个土壤样品上机测序的数据经过过滤、质控、去除嵌合体和段序列等操作后,共得到优质序列495 124条,平均每个样品有35 366条优质序列(表2)。通过Qiime软件按照97%相似度对所有优质序列进行聚类,一共得到5 821种OTU,平均每个样品中有2 640多个OTU。

表2 各样品最终序列数与OTU数统计Tab.2 Statistical table of final sequence numberand OTU number of each sample
2.2 土壤样品测序深度验证
稀释性曲线(Rarefaction curve)是从样本中随机抽取一定数量的个体,统计这些个体所代表的物种数目,并以个体数与物种数来构建曲线[15]。如图1所示,当曲线趋于平坦时,说明随着测序的加深获得的新物种的量较少。这就可以体现出测序的深度及测序的量合理性。

图1 样品稀释曲线Fig.1 Rarefaction curves for samples
同样,Shannon-Wiener曲线是利用各样本的测序量在不同测序深度时的微生物多样性指数构建曲线,如图2所示,当曲线趋于平坦时,说明数据量足够大,可以反映样本中绝大多数微生物的信息。
通过稀释曲线和Shannon-Wiener曲线,可以看出本次测序深度足够,当前测序结果可以反映出该样品中的土壤微生物的真实情况。
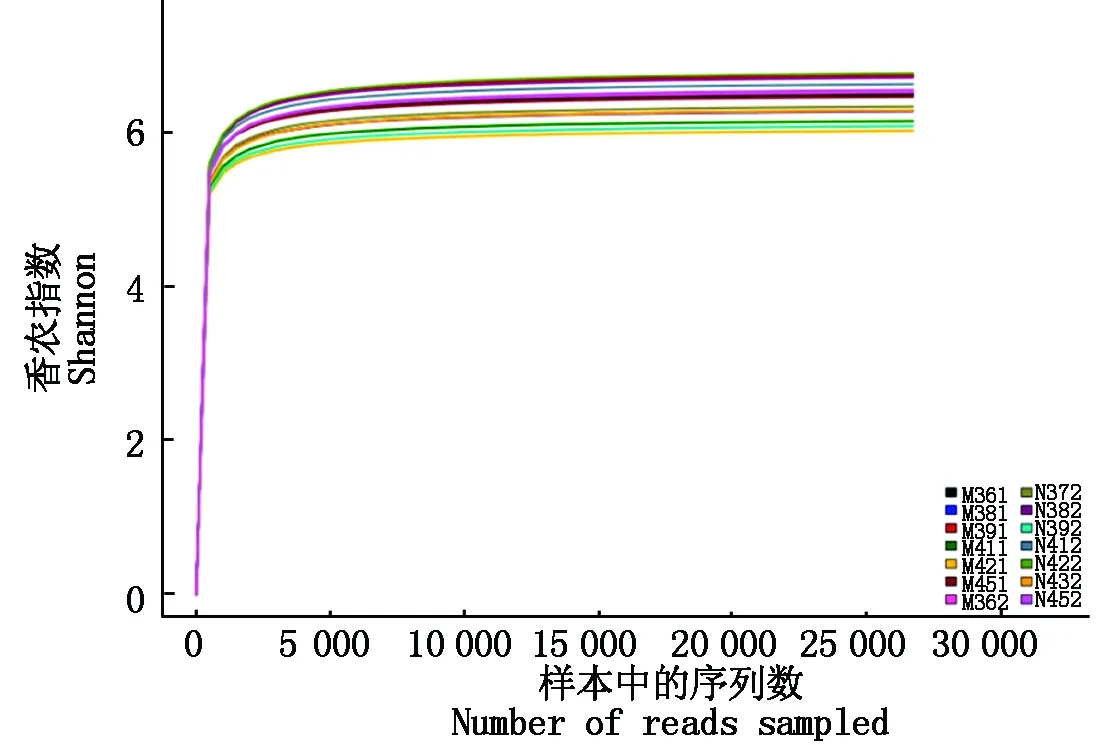
图2 Shannon-Wiener曲线Fig.2 Shannon-Wiener curves for samples
2.3 PCA分析及Anosim分析
PCA分析是一种对数据进行简化分析的技术。这种技术主要是对各个样品中的主成分进行分析。PCA分析可以反映出样品间的差异和相对距离[16]。如图3所示,大棚组(M1)与大田组(N1)的样品大部分集中在2个区域,并且两组间相对距离较远,说明两组间差异较大。N1组的几乎所有样品的相对距离都很近,说明大田的样品微生物组成相似度比较高,这可能是由于大田是开阔环境,与外界大环境交流频繁,所以相互之间的差异性较小。而M1组的几个样品相对分散,说明大棚样品间也存在一定的差异性,可能是由于大棚环境相对封闭,不可移动,以及长期施肥用药在土壤中的积累,都会对土壤中微生物组成产生很大的影响。
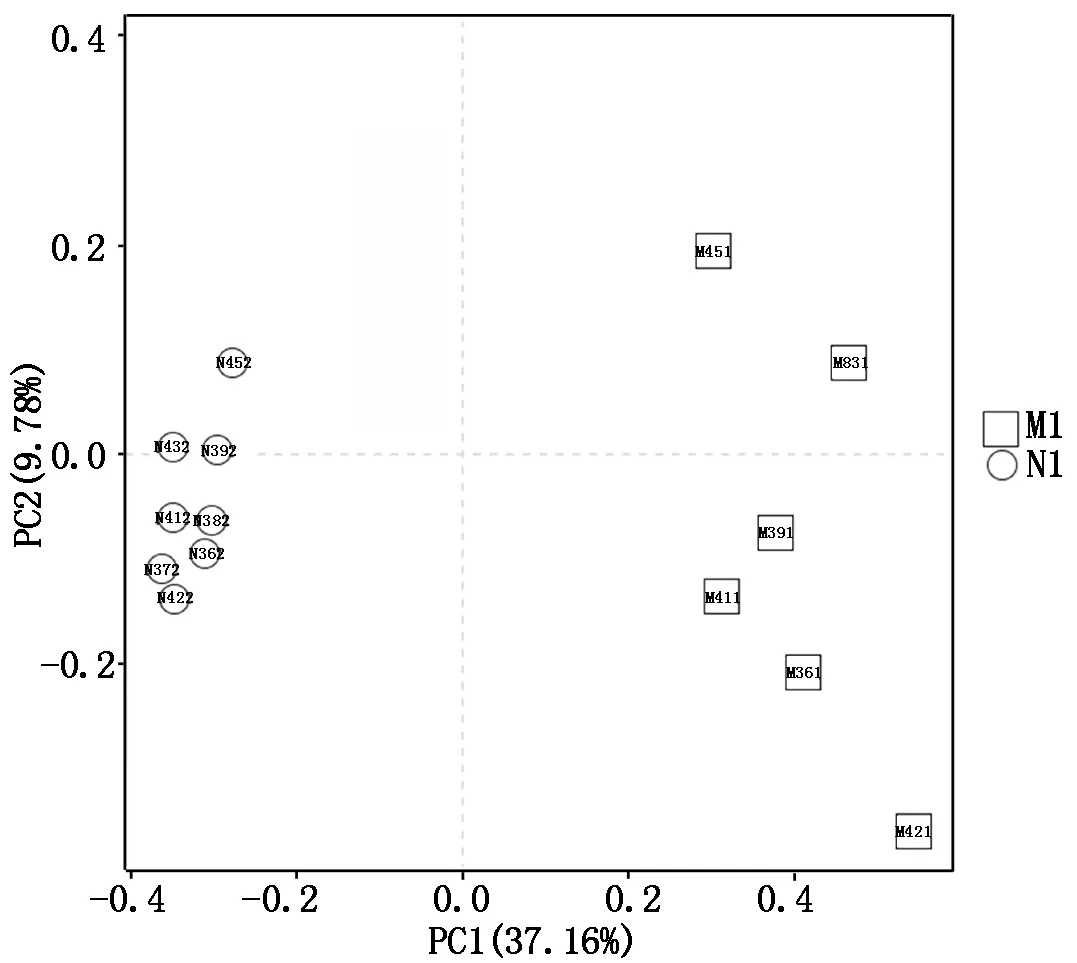
图3 基于OTU水平的PCA分析Fig.3 PCA analysis based on OTU level
Anosim相似性分析是用来检验组间的差异是否显著大于组内差异,从而判断分组是否有意义[17]。如表3所示,R-value介于(-1,1)之间,R-value大于0,说明组间差异显著。R-value小于0,说明组内差异大于组间差异。统计分析的可信度用P-value 表示,P< 0.05 表示统计具有显著性。表3中,大棚组与大田组的R-value是0.291 8大于0,说明这2组组间差异显著,而且P-value等于0.001远远小于0.05,说明这2组具有显著性差异。
通过PCA分析和Anosim分析,可以看出这2组样品间存在明显的差异,说明这样的分组是有意义的。接下来可以进一步对组间存在的差异进行分析。
表3 两组样品Anosim分析计算结果Tab.3 Anosim analysis and calculation results of two groups
2.4 两组样品的物种组成分析
2.4.1 门水平的物种组成分析 从门的水平来看(图4),2组样品中一共比对出已知菌门39种。其中,优势菌门以及在大棚组和大田组所占的比例分别是变形菌门(Proteobacteria,39.8%和38.2%)、放线菌门(Actinobacteria,14.4%和21.4%)、酸杆菌门(Acidobacteria,12.0%和15.4%)、拟杆菌门(Bacteroidetes,8.6%和6.9%)、绿弯菌门(Chloroflexi,7.9%和5.8%)、厚壁菌门(Firmicutes,4.5%和1.7%)和芽单胞菌门(Gemmatimonadetes,4.2%和3.7%)这7个细菌门类。在这7个优势菌门中,存在明显差异的有2个菌门。一个是放线菌门,在大田组中明显多于大棚组;另一个是厚壁菌门,在大田组明显少于大棚组。

图4 门水平物种组成Fig.4 Species composition in phylum
2.4.2 纲水平的物种组成分析 从纲水平来看(图5),2组样品中的优势菌纲一共有15种。这15种优势菌纲及其在M1和N1这2组中的比例分别是:α-变形菌纲(Alphaproteobacteria,19.4%和19.6%)、酸杆菌纲(Acidobacteria,10.6%和13.9%)、γ-变形菌纲(Gammaproteobacteria,10.6%和6.9%)、放线菌纲(Actinobacteria,8.1%和12.0%)、Δ-变形菌纲(Deltaproteobacteria,5.3%和6.2%)、β-变形菌纲(Betaproteobacteria,4.3%和5.4%)、芽单胞菌纲(Gemmatimonadetes,4.2%和3.7%)、纤维黏网菌纲(Cytophagia,3.7%和2.4%)、杆菌纲(Bacilli,3.5%和1.6%)、Anaerolineae(3.1%和1.3%)、鞘脂杆菌纲(Sphingobacteriia,3.1%和3.4%)、酸微菌纲(Acidimicrobiia,2.8%和3.4%)、嗜热油菌纲(Thermoleophilia,2.0%和3.6%)、黄杆菌纲(Flavobacteriia,1.7%和0.6%)和全噬菌纲(Holophagae,1.2%和1.4%)。
在上述15种优势菌纲中,经过统计学分析,大棚组与大田组相比相对丰度明显增高的是纤维黏网菌纲、杆菌纲和黄杆菌纲等,而相对丰度明显降低的是酸杆菌纲和β-变形菌纲等。

图5 纲水平物种组成Fig.5 Species composition in class
2.4.3 属水平的物种组成分析 进一步从属水平来看(图6),2组的优势菌属一共有6种,分别是节细菌属(Arthrobacter)、鞘氨醇单胞菌属(Sphingomonas)、德沃斯氏菌属(Devosia)、芽孢杆菌属(Bacillus)、黄单胞菌属(Xanthomonas)和溶杆菌属(Lysobacter)。其中,节细菌属在大田组(2.9%)的丰度明显多于大棚组(0.9%)。而在大棚组中明显增多的菌属有芽孢杆菌属、黄单胞菌属和溶杆菌属,它们在大棚组中的相对丰度分别是2.0%,1.1%,1.0%,在大田组中仅占1.0%,0.6%,0.3%。
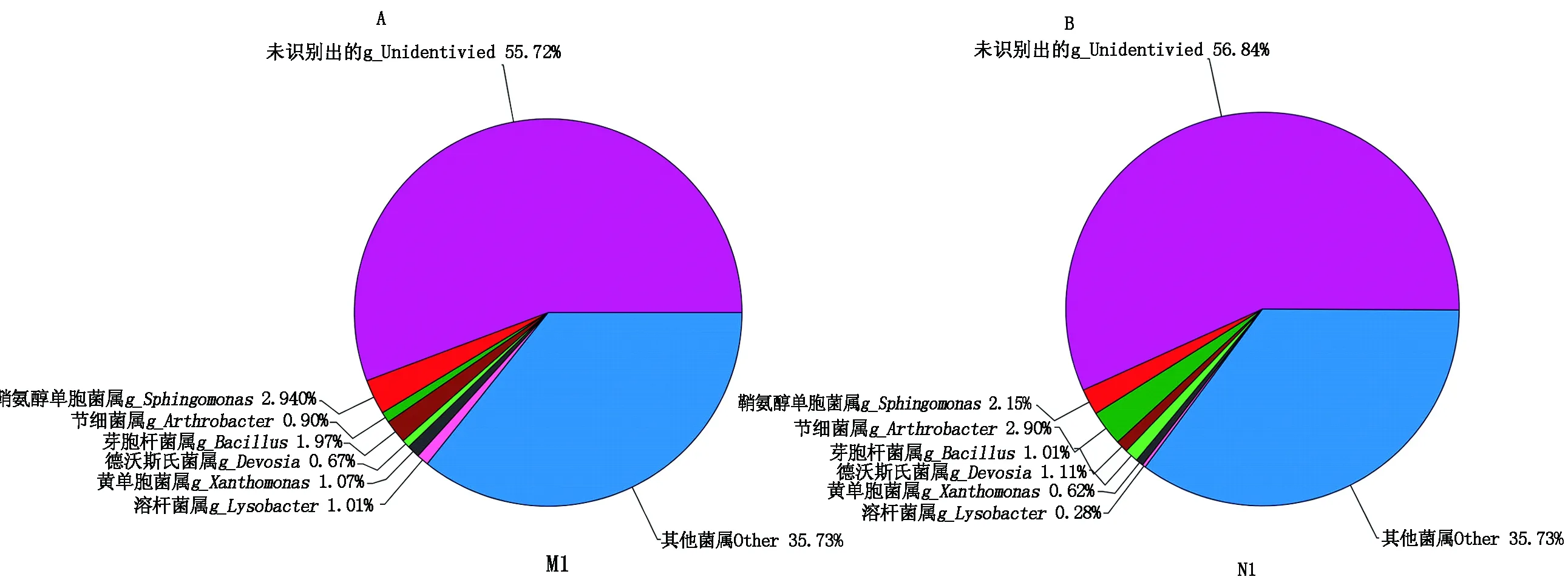
图6 属水平物种组成Fig.6 Species composition in genera of pie
另外,土壤中还有一些丰度较低的菌属,它们在2组中也存在着明显的差异。如表4所示,这些是2组间一部分差异较大的菌属及其可能的功能。从表4可以看出,大棚组中病原菌与可以产生抗菌物质的菌属都很多,这可能是由于大棚环境封闭,以及长期施肥用药导致土壤中重金属的积累,破坏了土壤微生物的平衡,容易富集一些病原微生物,同时相对应的土壤中拮抗的微生物也会多。而大田组中固氮、降解纤维素和降解重金属的微生物较多,这应该是由于大田与外界环境交流频繁,所以一些可以修复土壤和固氮的菌群相对丰度较大。
2.5 两组间LEfse分析
基于分类信息的LEfse分析的进化分支图(图7)可以更好地体现2组间存在的差异物种。从图7可以看出,浅色部分是大棚土壤中的优势微生物,其中浅色区域较大的是厚壁菌门和拟杆菌门;深色部分是大田土壤中的优势微生物,其中放线菌门的区域较大。这与之前的分析保持一致。LEfse分析的进化分支图更可以形象地看出各个优势菌门中具体哪些菌群是优势菌群。

表4 大棚组与大田组土壤微生物差异菌属比较分析Tab.4 Comparative analysis of soil microbial diversity in greenhouse and field groups

图7 基于分类信息的LEfse分析的进化分支Fig.7 Evolutionary branch graph of LEfse analysis based on classified information
2.6 Alpha多样性分析
Alpha多样性分析是通过对单样品的多样性分析反映微生物群落的丰度和多样性。常用的有:Chao1值,即菌种的丰富度指数,用来估计群落中的OTU数目,数值越大说明OTU数目越多;Shannon值,用来估算样品中微生物多样性的指数,同样也是数值越大说明群落多样性越高[28]。
从图8可以看出,大棚组的2个数值都要低一些,说明大棚比大田的微生物多样性要低一些。这应该是大棚较为封闭的原因,长期施肥用药以及高湿高温的环境,使得土壤中有害物质和重金属的积累破坏了土壤微生态的平衡,导致大棚土壤中微生物多样性较差。而大田环境开阔,与大环境交流频繁,土壤中微生物多样性较高,具有较好的修复能力。

图8 Alpha多样性指数boxFig.8 Alpha diversity index of box diagram
3 结论与讨论
本试验利用Illumina Miseq 测序平台对河北省中南部地区的大棚与大田土壤进行高通量测序,测定了土壤中细菌16S rDNA的V3-V4可变区的序列,并对其进行比对注释和多样性分析[29]。本次测序一共得到了495 124条优质序列,产生了5 821个OTU。通过对这些OTU进行注释,获得了土壤样品的多样性信息,并对其进行Alpha多样性分析和β多样性分析。通过Alpha多样性分析表明,大田土壤中的微生物多样性要多于大棚土壤,这可能是因为大田露天开阔的环境,使得大田土壤会随着风雨的变化与更多地区土壤微生物进行交流。而大棚相对封闭,与外界交流的范围和频率要少很多,且长期的高温高湿环境以及长期的施肥用药,导致土壤中有害物质和重金属积累,所以大田土壤比大棚土壤的细菌多样性要多。根据相关报道,土壤中微生物的多样性越多,土壤的自我修复能力以及抗病的能力就会越强。
通过物种组成图可以更进一步地了解大棚与大田土壤中哪些微生物有着明显的差异。从门水平来看,2组的优势菌群均为变形菌门、酸杆菌门、放线菌门和厚壁菌门等7个门类。其中,变形菌门所占比例最大,约占40%左右[30]。而放线菌门与厚壁菌门在2组中所占的比例就有所不同,差异最大的是放线菌门,在大棚中的丰度为14.4%,而在大田中为21.4%。因为许多放线菌会产生抗生素,所以这可能也是大田比大棚抗病性更强的原因。从纲水平的物种组成图可以看出,2组土壤样品的优势菌群基本相似,一共有15种优势菌群。同样属于变形菌门的几个纲所占比例较大。从属水平来看,2组的优势菌群一共有6种,节细菌属(Arthrobacter)、鞘氨醇单胞菌属(Sphingomonas)、德沃斯氏菌属(Devosia)、芽孢杆菌属(Bacillus)、黄单胞菌属(Xanthomonas)和溶杆菌属(Lysobacter)。其中,节细菌属在2组中差异较大,大田中要比大棚中多很多。节细菌属在土壤中具有固氮和降解重金属的作用。这可能是大田土壤在固氮能力和修复能力方面要比大棚土壤强的一个原因。一些相对丰度低的菌属中,大田土壤比大棚土壤相对丰度较多的也是一些具有固氮作用和降解纤维素作用的菌属。这也验证了,大田在固氮能力和自我修复的能力上比大棚土壤要强。
综上所述,通过对河北南部地区的大棚土壤与大田土壤进行高通量测序,初步了解了2组样品中细菌的组成情况。2组土壤中优势菌群种类基本相似,但有些菌群的相对丰度差异较大。从门和纲的水平来看,大田中的放线菌的相对丰度都是高于大棚土壤,而大棚中的厚壁菌门要多一些。从属水平优势菌属看,具有固氮和降解重金属的节细菌属在大田中的相对丰度要高于大棚。这可能是由大棚相对封闭的高温高湿环境以及长期施肥用药引起的重金属积累所导致的。这就使得大田的细菌多样性高于大棚,而且大田土壤中在抗菌方面、固氮方面与修复方面的菌群也要多于大棚土壤。土壤中的微生物彼此之间存在着千丝万缕的联系[31],或许以后可以通过移植土壤来改善大棚中的种植环境,提高产量和抗病能力。
