基于家蚕中肠RNA-seq数据的新基因发掘及初步分析
2018-08-27周凯月李玉霞郝长富徐安英
周凯月, 唐 健, 李玉霞, 郝长富, 徐安英, 2*
(1.江苏科技大学生物技术学院,江苏镇江 212018;2.中国农业科学院蚕业研究所,江苏镇江 212018)
我国是茧丝绸生产和出口大国,家蚕作为重要的吐丝昆虫,具有很高的经济价值,在生物反应器和模式生物中也显现出广阔的应用前景。随着家蚕研究的深入,越来越多的新基因被发掘出来,一些新基因往往发挥着极为重要的作用,甚至是某些疑难问题的突破点,人们对新基因功能的研究也越来越重视。
转录组研究是一个发掘功能基因的重要途径,与基因组学相比,转录组学只研究被转录的基因,研究范围缩小,针对性更强[1],越来越多地被运用到基因功能的相关研究中。转录组是特定组织或细胞在某一发育阶段或功能状态下转录出来的所有RNA的总和[2],主要包括mRNA和非编码RNA(ncRNA)。转录组测序即通过第二代高通量测序技术对特定组织或细胞的转录产物(主要是全部mRNA)反转录后测序并对其进行生物信息学分析的技术[3],是当前在全基因组水平上研究基因表达模式的主要技术[4],目前该技术已被广泛应用于生物信息学研究的多个领域[5]。笔者拟采用RNA-seq技术对所构建的家蚕中肠的转录组进行测定,并在基因组水平上进行转录组分析,对新基因进行初步发掘,以期为新基因的功能鉴定基础。
1 材料与方法
1.1材料试验所用家蚕品种为抗BmNPV家蚕品种QFN和常规品种QF,来源于中国农业科学院蚕业研究所蚕资源中心课题组。
1.2方法
1.2.1转录组测序。首先进行样品检测、构建RNA文库,文库质控合格后,采用HiSeq2500进行高通量测序, Illumina HiSeq2500高通量测序获得Reads或碱基信息,筛选除去冗余后得到Clean Reads,通过solexa QA软件对其进行质量检测可得到高质量的Clean Reads[6]。
1.2.2转录组数据比对。对于Clean Reads需要用高效的序列比对软件TopHat2将其与参考基因组进行序列比对,得到Mapped Reads。比对效率可以直接反映出转录组数据的利用率[6]。
1.2.3新基因分析。通过Cufflinks软件对Mapped Reads进行组装,将得到的序列与参考基因组注释信息进行比对,寻找未知的新基因。再利用Blast软件对新基因进行功能注释,然后利用各个数据库分别对新基因的NR注释信息、COG功能注释及其分类、KEGG注释通路进行分析,获得新基因的相关注释信息。
1.2.4部分新基因的克隆分析。以家蚕抗性品种QFN的中肠组织cDNA为模板,随机挑选3个新基因(Silkworm New Gene 1、Silkworm New Gene 5、Silkworm New Gene 20)设计引物,进行RT-PCR扩增。PCR反应体系为(25 μL):1 μL cDNA模板,2.5 μL 10×PCR Buffer,2 μL 10 mmol/mL dNTP,1 μL 20 pmol/mL上下游引物,0.3 μL 5 U/μL ExTaqDNA聚合酶,加双蒸水补充至25 μL。PCR反应后检测扩增片段的大小,与目的片段大小是否相符。所需引物序列见表1。
采用SanPrep柱式DNA胶回收试剂盒进行目的片段的回收和纯化。试验前确认Wash Solution中是否加入乙醇,将胶回收纯化的PCR产物连接到pMDTM18-T Vector上,用冷热刺激的方法转化感受态细胞,过夜培养后挑取独立菌落进行培养,再进行菌液PCR扩增判断是否为目的条带,挑选阳性菌液送生工生物工程(上海)股份有限公司测序鉴定。
2 结果与分析
2.1测序数据为研究家蚕感染BmNPV后蚕体内基因表达调控情况,以家蚕抗性品种QFN和常规品种QF中肠组织为材料进行转录组测序分析(表2)。参考基因组组装能否满足信息分析的需求,可以通过转录组数据与参考基因组序列的比对结果评估(表3)。
经筛选,2个文库共获得12.8 Gb Clean Data,QF、QFN的≥Q30的碱基百分比分别为86.03%、87.01%。

表1 部分新基因RT-PCR引物序列
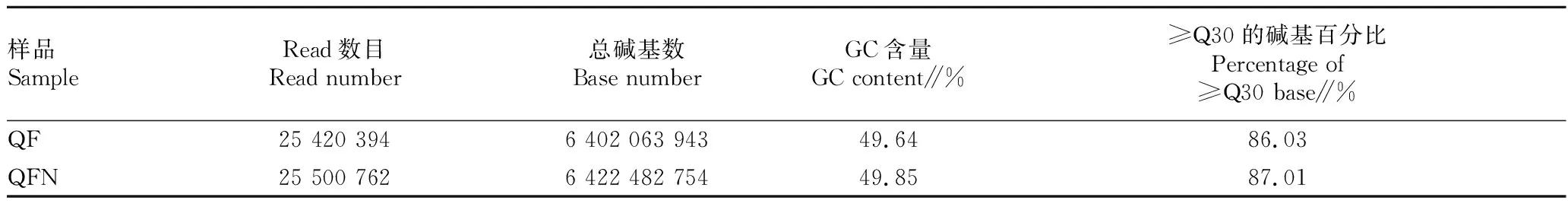
表2 样品测序数据统计

表3 Clean Data与参考基因组比对结果统计
文库中比对到参考基因组上的Reads在Clean Reads的效率达69.36%和70.24%,其中比对到参考基因组唯一位置的Reads在Clean Reads中所占比例分别为58.66%、60.75%。
2.2新基因分析
2.2.1新基因发掘及基因结构的分析。对测序得到的序列进行拼接和组装,与原有的一些基因组注释信息进行比对,寻找未被注释的新基因。该研究过滤掉编码的肽链过短(少于50个氨基酸残基)或只包含单个外显子的序列,得到了788个新基因。如Silkworm New Gene1007位于nscaf2794基因序列172~4 311的正链上,包含5个外显子;Silkworm New Gene1008位于nscaf2795基因序列2 402 828~2 404 107的正链上,包含3个外显子,部分新基因的文件见表4。
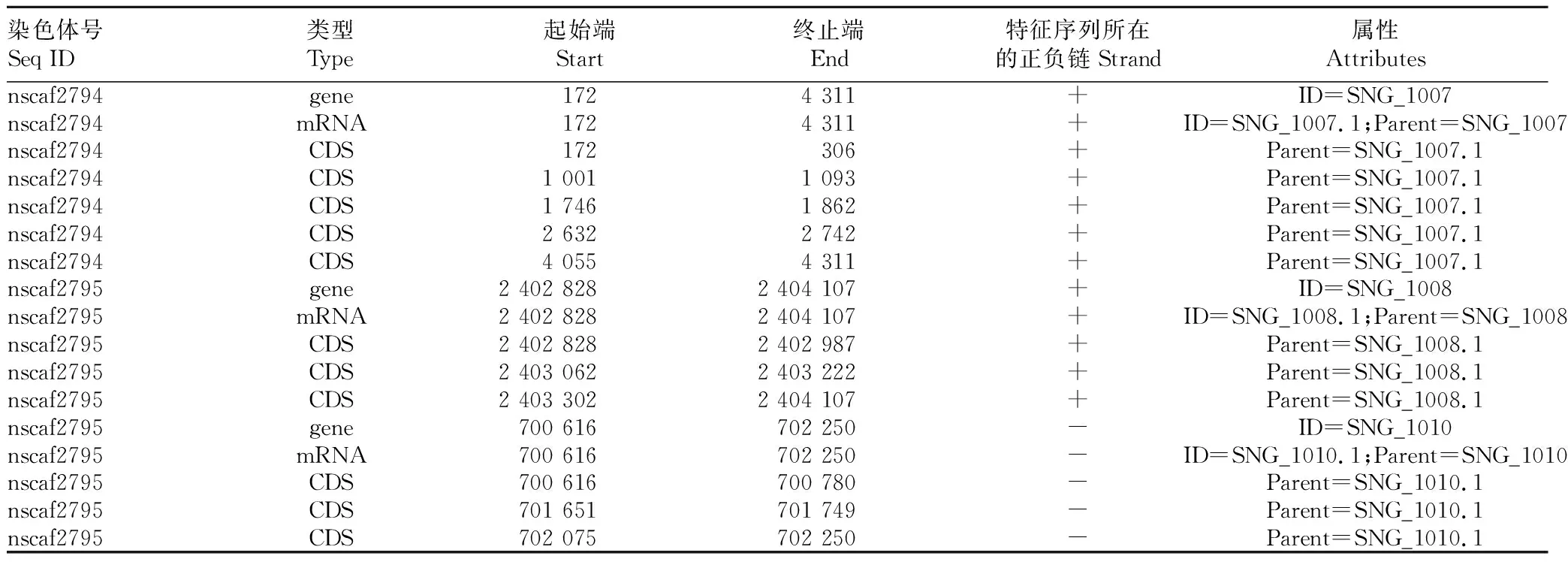
表4 部分新基因的文件
注: SNG为Silkworm New Gene简写
Note: SNG is short for silkworm new gene
2.2.2新基因功能注释。使用Blast软件将发掘的新基因分别同NR[7],Swiss-Prot[8]、GO[9]、COG[10]、KEGG[11]数据库进行序列比对[12],结果发现788个新基因中746个得到了注释,各数据库得到注释的基因数分别为746、418、443、187、224个。新基因功能注释结果统计详见表5。

表5 新基因功能注释结果统计
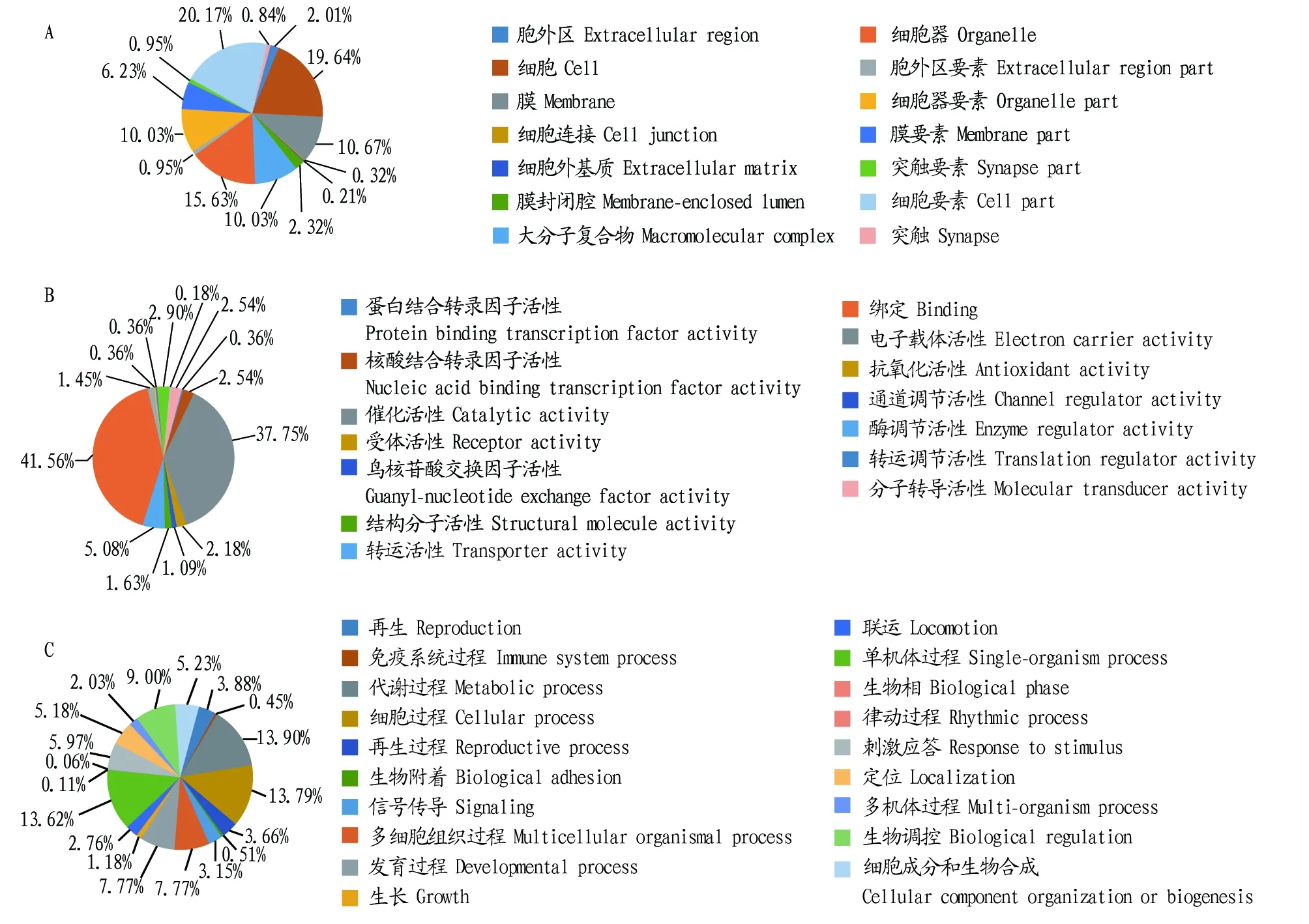
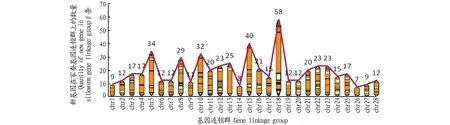
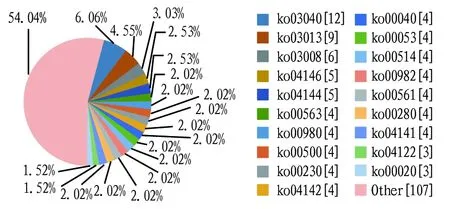
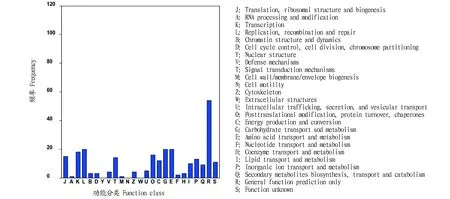
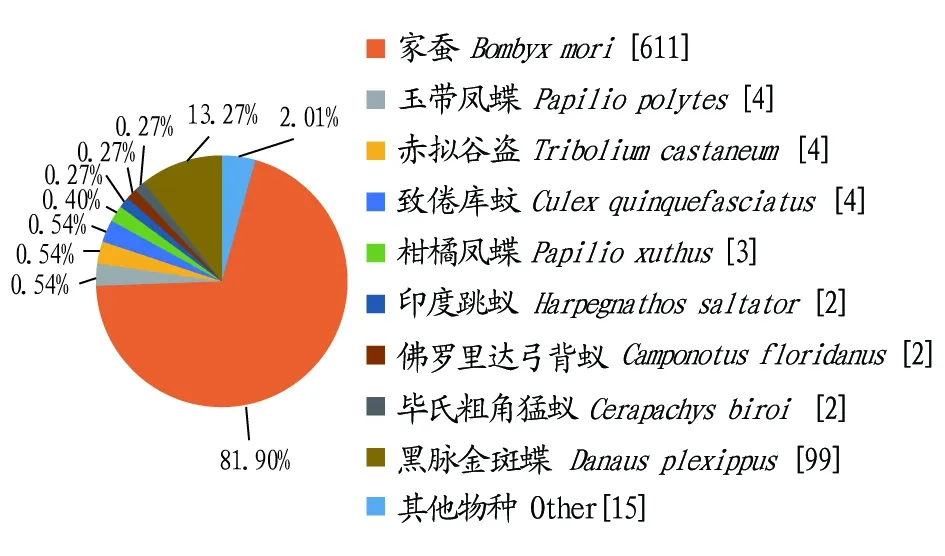
同时发现,其中746(94.67%)个新基因与数据库中匹配到的序列具有显著的相似性(E 图1 注释基因E值分布Fig.1 Distribution of E-value for annotated genes 2.2.3与注释基因匹配的物种分布。利用BlastX将组装出来的unigene序列与NR数据库进行比对后,共找到746个unigene与其他近缘生物的已知基因具有不同程度的同源性,746个注释的基因中,有611条(81.90%)基因与家蚕(Bombyxmori)序列同源,99条(13.27%)与黑脉金斑蝶(Danausplexippus)序列同源、4条(0.54%)与玉带凤蝶(Papiliopolytes)序列同源、4条(0.54%)与赤拟谷盗(Triboliumcastaneum)序列同源、4条(0.54%)与致倦库蚊(Culexquinquefasciatus)序列同源、3条(0.40%)与柑橘凤蝶(Papilioxuthus)序列同源、2条(0.27%)与印度跳蚁(Harpegnathossaltator)序列同源、2条(0.27%)与佛罗里达弓背蚁(Camponotusfloridanus)序列同源、2条(0.27%)与毕氏粗角猛蚁(Cerapachysbiroi)序列同源,仅有15条(2.01%)与其他物种序列相匹配(图2)。 2.2.4新基因GO富集分析。利用Blast2Go软件对筛选到的基因进行GO富集分析,结果显示,基因主要注释到细胞组分、分子功能和生物学过程3个分支中,分别有947、551和1 777个(图3)。在细胞组分模块中(图3A),注释到细胞(cell)、细胞部分(cell part)的基因数目较多,分别占19.6%和20.0%;在分子功能模块中(图3B),注释到黏合(binding)和催化活动(catalytic activity)的基因数目较多,分别占41.0%和37.7%。在生物学过程模块中(图3C),注释到细胞过程(cellular process)和单组织过程(single organismprocess)及代谢过程(metabolic process)的基因数目较多,分别占13.8%、13.6%和13.9%。 图2 与注释基因匹配的物种分布Fig.2 Distribution of species match to the annotated genes 2.2.5新基因在家蚕基因连锁群上的分布。将新基因的Locus与家蚕基因连锁群进行比对发现,有545个新基因分布在不同的染色体上,且在28条染色体上都有分布(图4),在18号染色体上分布数量最多,有58个,其中在nscaf2902上分布有54个;在15号染色体上分布有40个,其中在nscaf2888上分布有36个;在26号染色体上分布最少,仅有7个。 2.2.6新基因通路富集分析。通过KEGG分析,对新基因进行通路富集分析发现,共有 198个新基因注释到KEGG数据库中,分布于85条已知的通路中。映射基因最多的5个通路分别为剪接体(splicesome)(ko03040、12条)、RNA转运(RNA transport)(ko03013、9条)、真核细胞核糖体合成(ribosome biogenesis in eukaryotes)(ko03008、6条)、过氧物酶体(peroxisome)(ko04146、5条)、内吞(endocytosis)(ko04144、5条)。映射到的信号通路见图5。 2.2.7新基因COG数据库功能注释。将新基因与COG数据进行比对,并进行功能注释与分类,结果发现共有22个类别里的258个新基因得到了注释(图6),其中,一般功能(general function prediction only)的基因占总体的20.93%,所占比例最大;复制、重组和修复(replication, recombination and repair )、碳水化合物的运输和代谢(carbohydrate transport and metabolism)、氨基酸的运输和代谢(amino acid transport and metabolism)3个类别共居第2位,占总体的7.75%;转录(transcription)占总体的7.36%,其余分类的基因数较少,其中核结构(nuclear structure)、细胞运动(cell motility)、真核细胞的细胞外结构(extracellular structures)3个类别里均无新基因出现。 2.2.8部分新基因的克隆分析。以秋丰N中肠组织的cDNA为模板,随机挑选3个新基因设计引物扩增目的基因,获得特异性片段,与预期片段大小相符,经测序结果与参考序列比对后发现编码序列高度相似。 该研究基于所选参考基因组序列,共发掘788个新基因,通过生物信息学软件将发掘的新基因与NR、Swiss-Prot、GO、COG及KEGG数据库进行序列比对,共获得746个新基因的注释信息;746个注释的基因中,其中611条(81.90%)基因与家蚕序列同源;利用Blast2Go软件对筛选到的基因进行GO富集分析,结果显示,基因主要注释到细胞组分、分子功能和生物学过程3个分支中,分别有947、551和1 777个,注释到细胞部分(cell part)、黏合(binding)及代谢过程(metabolic process)的基因数目最多;将新基因的Locus与家蚕基因连锁群进行比对发现,有545个新基因分布在不同的染色体上,且在28条染色体上都有分布;通过KEGG分析对新基因进行通路富集分析发现,共有 198个新基因注释到KEGG数据库中,分布于85条已知的通路中;将新基因与COG数据进行比对,并进行功能注释与分类,结果发现共有22个类别里的258个新基因得到了注释。新基因与各个数据的序列比对结果,进一步证实了新基因的存在,该研究对新基因的功能做了初步分析,关于新基因具体的功能还需要进一步研究。 注:A.细胞组分;B.分子功能;C.生物学过程Note:A.Cellular components;B.Molecular function;C.Biological processes图3 注释基因在GO中的分类情况Fig.3 Classification of annotated genes in GO 图4 新基因在家蚕基因连锁群的分布Fig.4 Distribution of new gene in silkworm gene linkage group 图5 KEGG通路富集分布Fig.5 KEGG pathways distribution 图6 COG数据库中功能注释的unigenes分类Fig.6 COG function classification of unigenes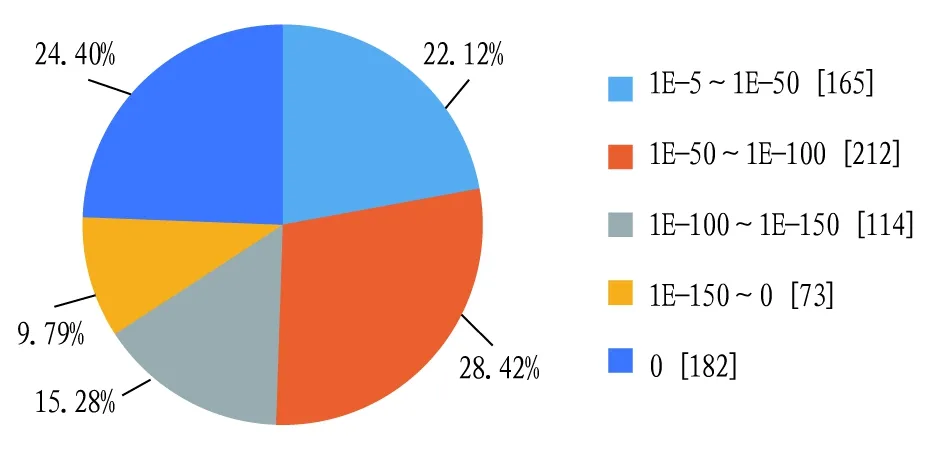
3 讨论