引发剂用量对环状对苯二甲酸丁二醇酯聚合反应动力学及产物的影响
2018-08-21苏银河王新威于俊荣胡祖明
苏银河, 王新威, 于俊荣, 王 彦, 诸 静, 胡祖明
(1.东华大学 纤维材料改性国家重点实验室, 上海 201620;2. 上海化工研究院 上海市聚烯烃催化技术重点实验室, 上海 200031)
环状对苯二甲酸丁二醇酯(CBT)为不同低分子量大环寡聚酯结构的混合物[1],加热到190 ℃熔融后,其黏度约为0.020 Pa·s,与水在20 ℃下的黏度(0.001 Pa·s)接近,流动性非常好,加入引发剂后其可在适当的温度下开环聚合生成聚环状对苯二甲酸丁二醇酯(pCBT),聚合时间相对较短,聚合过程基本无热量(焓变ΔH非常小)和挥发性有机化合物(VOC)的释放。由于CBT树脂润湿能力强、填充能力强、加工黏度低,与各种填料、增强材料和高分子材料的相容性好,CBT树脂有望成为制备纤维增强复合材料的理想基体[2-3]。
CBT的开环聚合反应可以通过改变反应温度及引发剂的用量进行调控,使得CBT的聚合时间控制在几十秒至几十分钟之内。在CBT聚合成为pCBT过程中,体系黏度由小变大,体系发生相变,从液态转变为固态。CBT树脂的黏度对其加工性能有极大的影响,因此,掌握其在聚合反应过程中熔体黏度的变化和相转变是非常重要的。针对高于pCBT结晶温度的CBT聚合反应动力学已有研究人员进行了细致研究[4],而对低于或接近于pCBT结晶温度的CBT聚合反应动力学和引发剂用量对聚合产物结构性能影响的研究未见报道。
本文利用流变学的方法研究CBT的聚合反应动力学,系统地考察引发剂加入量对CBT树脂开环聚合过程及产物分子量和分布的影响,并对pCBT的结晶结构、热性能和力学性能做详细研究。
1 试验部分
1.1 试验原料
CBT,购于美国Cyclics公司,牌号CBT100,重均相对分子质量Mw=220ng/mol,n=2~7(n为CBT的聚合度)。
二羟基丁基氯化锡,引发剂,购于阿达玛斯公司,牌号Fascat 4101,纯度98%。
丙酮、四氢呋喃、苯酚和四氯乙烷均购于国药集团化学试剂有限公司,分析纯。
1.2 pCBT样品制备
将CBT100溶解于丙酮中,加入一定量的二羟基丁基氯化锡,搅拌溶解,使引发剂与CBT充分混合,然后在室温下挥发掉大部分的溶剂,最后将混合物放入真空干燥箱内,80 ℃干燥至恒重。按加入引发剂的质量分数(0.10%、 0.25%、 0.50%和1.00%)分别将相应的混合物标记为CBT-0.10、 CBT-0.25、 CBT-0.50和CBT-1.00。
采用TA公司AR 2000型旋转流变仪进行CBT的原位聚合流变测试,平行板夹具直径为25 mm。先将流变仪温度升至190 ℃,随后将干燥的CBT添加至平行板之间,快速调节两板间距为1 mm,之后对熔体进行动态流变测试直至熔体黏度不再发生变化,测试频率为1 rad/s,应变为1%。检测等温条件下不同引发剂用量的CBT开环聚合过程,最终得到产物pCBT。聚合测试过程中使用氮气作为保护气。
2 测试与表征
示差扫描量热(DSC)测试。采用美国TA仪器公司Q 20型差示扫描量热仪进行DSC分析。称取5~10 mg CBT样品,在氮气条件下以10 ℃/min的升温速率从室温升至250 ℃,恒温5 min消除热历史,之后以10 ℃/min 的速率降温至室温,恒温5 min,再次以10 ℃/min速率升温至250 ℃,记录整个升降温过程中热焓随温度的变化关系。测试pCBT热性能时第一次升温速率为100 ℃/min,其余升温程序不变。计算样品结晶度时采用100%结晶的PBT的熔融热焓,ΔH0=144.5 J/g[5]。
凝胶色谱(GPC)测试。采用美国Waters公司的HT 3-515型凝胶色谱仪进行pCBT的分子量及其分布测试。测试温度为30 ℃,提洗液为六氟异丙醇,流速为1.0 mL/min,样品质量浓度为1.0 mg/mL,测试时间为20 min。
特性黏度([η])测试。采用毛细管直径为0.7~0.8 mm的乌氏黏度计进行样品的特性黏度测定,以苯酚-四氯乙烷(质量比6 ∶4)混合液为溶剂,测试温度为30 ℃。采用一点法(式(1))计算出样品的特性黏度[η][6]。
(1)
式中:c为溶液浓度,本试验采用的溶液质量浓度为5 g/L;t为样品溶液流出时间;t0为纯溶剂流出时间。根据Mark-Houwink公式[7](式(2))求得样品的黏均分子量Mη。
(2)
聚合转化率(C)测试。取小片样品称重,放入40 ℃四氢呋喃(THF)中浸泡24 h,以去除没有参加反应的CBT。然后取出样品放入80 ℃真空烘箱内,烘干至恒重。CBT的聚合转化率计算如式(3)所示。

(3)
式中:m为经THF溶解后样品的质量;m0为原始样品的质量。
X射线衍射(XRD)分析。采用日本Rigaku公司的D/max-2550 PC型X射线衍射仪进行样品衍射图谱测定,2θ扫描范围为5°~60°进行扫描,Cu靶Kα辐射,扫描速度为2(°)/min,采用软件Jade 5.0分峰处理后求得各试样的结晶度,并由谢乐公式计算各晶面法线方向的晶粒尺寸。
力学性能测试。将聚合得到的pCBT在微型注塑机上注塑成标准样条,采用美国Instron公司Instron 5567型试验机分别进行样条的拉伸和弯曲性能测试,每个样品测试5次取平均值。拉伸性能测试标准为ISO 527,拉伸速率为50 mm/min;弯曲性能测试标准为ISO 178,压头速率为2 mm/min;冲击性能测试按照ISO 180:2000在多功能组合冲击试验机上进行。
3 结果与讨论
3.1 CBT树脂的DSC分析
CBT-0.50的第一次升温和降温、第二次升温DSC曲线如图1所示。由图1可知,第一次升温DSC曲线分别在115.9、 140.6、 155.8和187.8 ℃处出现了4个大小不一的吸热峰,对应了不同聚合度CBT的熔融行为,可说明CBT树脂是由不同相对分子质量的环状低聚物组成的。当温度超过其中任一组分的熔点时,CBT即发生开环聚合反应,生成线性大分子pCBT,当生成的聚合物分子量足够大时便会发生结晶行为,使得曲线在200.0 ℃左右出现一个较宽较弱的结晶放热峰,即在226.6 ℃处的吸热峰就是升温过程中所形成的pCBT结晶的熔融吸热峰。
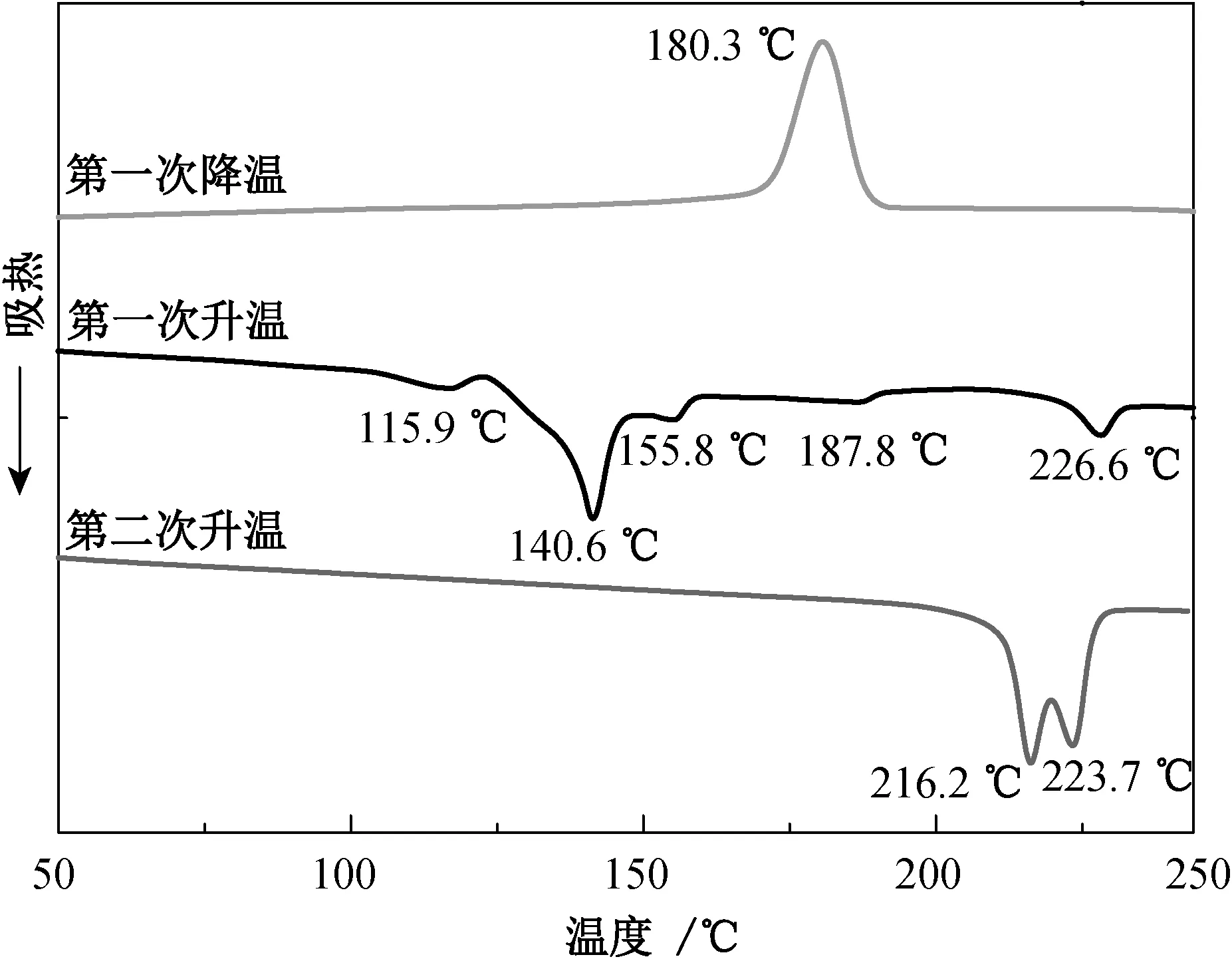
图1 CBT-0.50的第一次升温和降温、第二次升温DSC曲线Fig.1 First heating and cooling,and second heating DSC thermograms of CBT-0.50
在pCBT熔体的降温过程中,仅在180.3 ℃处出现一个单一的pCBT的结晶放热峰,并没有出现CBT的结晶峰。在第二次升温过程中,CBT的熔融吸热峰也全部消失,说明经过短时间的高温加热,CBT已全部转化为pCBT。第二次升温曲线在216.2和223.7 ℃处出现两个明显的熔融吸热峰,这是因为pCBT的重结晶行为造成的。这种重结晶行为是常规PBT也具有的结晶行为[8]。
3.2 CBT聚合过程研究
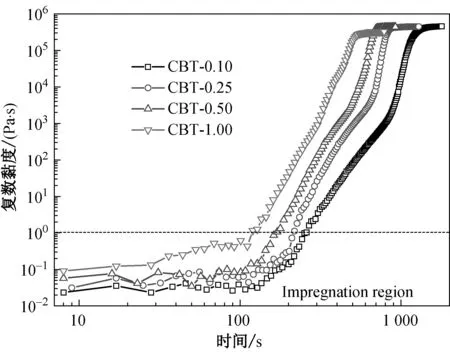
选择在低于pCBT的熔点温度条件下进行CBT的聚合,不同引发剂含量的CBT在190 ℃条件下开环聚合过程中熔体复数黏度和相位角随时间的变化分别如图2所示。
(a)复数黏度

(b) 相位角
图2不同引发剂含量CBT在190℃聚合过程中复数黏度和相位角随时间变化的曲线
Fig.2VariationofcomplexviscosityandphaseanglewithtimeforCBTpolymerizationat190℃withdifferentinitiatorcontents
CBT的开环聚合反应过程可以分为3个区域。区域Ⅰ是复数黏度曲线上的低黏度区,在这期间不同引发剂含量的CBT逐渐发生熔融但未开始发生链增长反应,即为聚合诱导期,熔体复数黏度小于1 Pa·s,这与文献[9]报道相符。区域Ⅰ内的熔体相位角在90°附近产生较大波动,这是由于CBT中高熔点组分还没有完全熔融[10]。聚合诱导期结束之后熔体进入黏弹转变区(区域Ⅱ),熔体复数黏度短时间内突增,从1 Pa·s迅速增加至105Pa·s以上,伴随着相位角开始骤降。区域Ⅱ内熔融状态下的CBT单体在引发剂作用下迅速发生开环聚合反应,pCBT聚合体分子量增大到一定程度,开始逐步结晶而引起相变。此时熔体体系储能模量与损耗模量相交,出现凝胶点(Tgel),发生熔体从黏性到弹性的转变。之后进入熔体弹性区(区域Ⅲ),此区域内CBT的聚合过程伴随着pCBT的结晶过程,体系弹性逐步增大,伴随着相位角迅速下降接近0°,这说明体系中pCBT的分子量逐步增大,并逐步结晶至结晶平衡。区域Ⅲ结束后熔体黏度到达高黏度平台区,此时,熔体体系黏度基本不再变化,接近弹性固体状态,相位角下降至0°左右。
由图2可知,无论引发剂用量多少,熔体复数黏度曲线均存在由低黏度平台区(区域Ⅰ)逐渐向高黏度平台区(区域Ⅲ)转变的过程。低黏度区CBT熔体黏度小于1 Pa·s,非常有利于浸润填料。不同引发剂用量条件下CBT开环聚合各阶段所用时间如表1所示。随引发剂用量的增加,低黏度平台区变小,且体系从低黏度平台区到达高黏度平台区所需时间也随之缩短。综上可以看出,引发剂的用量对于聚合反应速率的影响非常大,引发剂用量越多,反应越迅速,聚合完成时间越短。

表1 不同引发剂用量对CBT 在190 ℃开环聚合反应各区间完成时间的影响
3.3 pCBT分子量和反应产率分析
不同引发剂用量条件下CBT开环聚合所得pCBT的GPC曲线如图3所示。由图3可知,4条曲线均出现了一个对称的单峰,且无小分子CBT峰出现,表明聚合非常完全。引发剂用量变大时,试样的峰值对应的保留时间逐渐增加,pCBT分子量逐渐减小。由GPC测试所得的各pCBT的分子量及其分布(数均分子量Mn、重均分子量Mw和多分散性指数PDI)如表2所示,同时,表2还列出了不同引发剂用量条件下CBT的转化率及由黏度法测得pCBT的特性黏度[η]和黏均分子量(Mη)。由表2可知:CBT转化率均可以达到97%以上;随引发剂用量增加,产物Mn和Mw均明显下降,同时,其PDI呈下降趋势;由黏度法测得的pCBT的特性黏度和Mη也随着引发剂用量的增加而减小,该结果与GPC测试所得的数据相符合。这是由于较多的引发剂会产生较多的活性中心,与活性中心相连的分子链长度变小,分子量降低,反映在特性黏度上就表现为黏度下降。
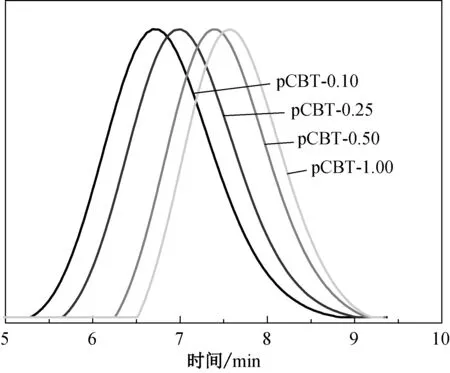
图3 不同引发剂用量条件下制得pCBT的GPC曲线Fig.3 GPC graph of pCBT with different initiator contents
3.4 pCBT的热性能和结晶性能分析
不同引发剂用量条件下制得的pCBT熔体的DSC曲线如图4所示,相关参数列于表3中。由表3可知,随着引发剂用量的增加,pCBT的结晶温度(Tc)升高、最终熔融温度(Tm)和结晶度(XC, DSC)等均呈上升趋势,并且pCBT的熔融双峰之间的温度差值也逐渐变大。产生熔融双峰的原因是不完善的晶体在熔融过程中发生二次结晶和重组。聚合物分子量越小则其分子链活动能力越强,降温结晶时,更容易排入晶区,使其结晶温度升高,结晶度变大。然而由于低分子量pCBT结晶速度过快,分子链来不及整齐排列,导致其晶型不完整,晶体尺寸分布较宽,在二次熔融时更易出现重结晶现象,产生熔融双峰,因此pCBT分子量越小,其DSC曲线上的熔融双峰越明显,两峰温差也越大。通常来说,聚合物的分子量越大,结晶熔点越高,但本文的试验结果与此相反,这可能是由于分子量小时,pCBT更易结晶。
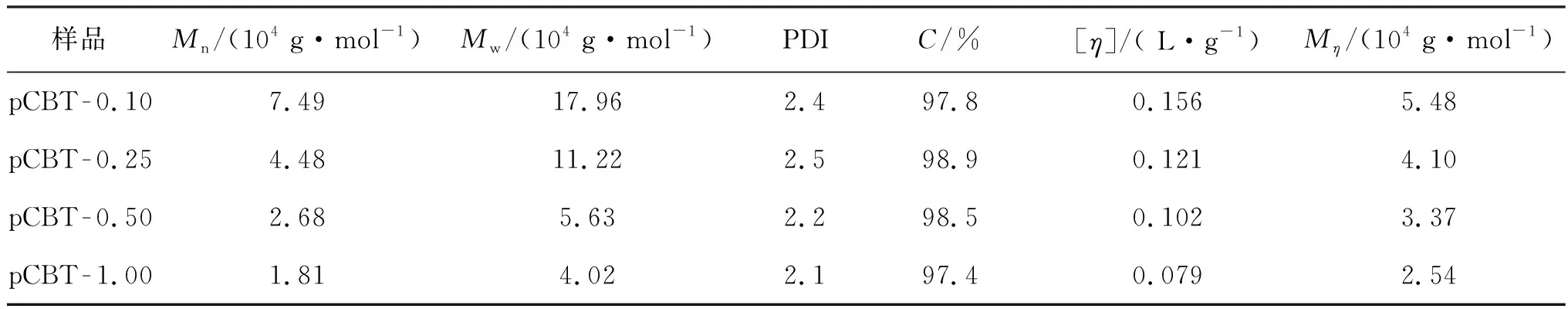
表2 引发剂用量对CBT转化率、pCBT分子量和特性黏度的影响Table 2 Effect of initiator content on the C of CBT, molecular weights and intrinsic viscosity of pCBT


(a) 降温结晶 (b) 二次升温

样品Tc/℃Tm1/℃Tm2/℃XC, DSC/%L011/nmL010/nmL110/nmL100/nmL111/nmXC, XRD/%pCBT-0.10181.3218.9221.330.98.587.026.095.835.3331.0pCBT-0.25185.0218.6223.935.28.536.515.335.235.0233.2pCBT-0.50187.8217.1224.937.36.576.255.935.854.5036.7pCBT-1.00189.6216.9226.040.16.035.894.054.574.3856.8
不同引发剂用量条件下CBT聚合所得各pCBT的XRD谱图如图5所示,其中出现的5个衍射峰依次对应于PBT三斜晶系中(001)、(010)、(110)、(100)和(111)晶面的特征衍射[11]。随着引发剂用量的增大,各衍射峰的峰强明显增强,pCBT结晶度逐渐增大,而各晶面法线方向的晶粒尺寸(L011、L010、L110、L100和L111)逐渐减小(如表3所示)。由此说明,随着引发剂用量的增大,pCBT自由活性链变多,体系中结晶成核密度增加,从而导致体系中微晶尺寸变小,结晶度增大。同时,较小的分子量也会促进pCBT的结晶,导致其结晶度上升,与DSC法测得的结晶度数据趋势相符。
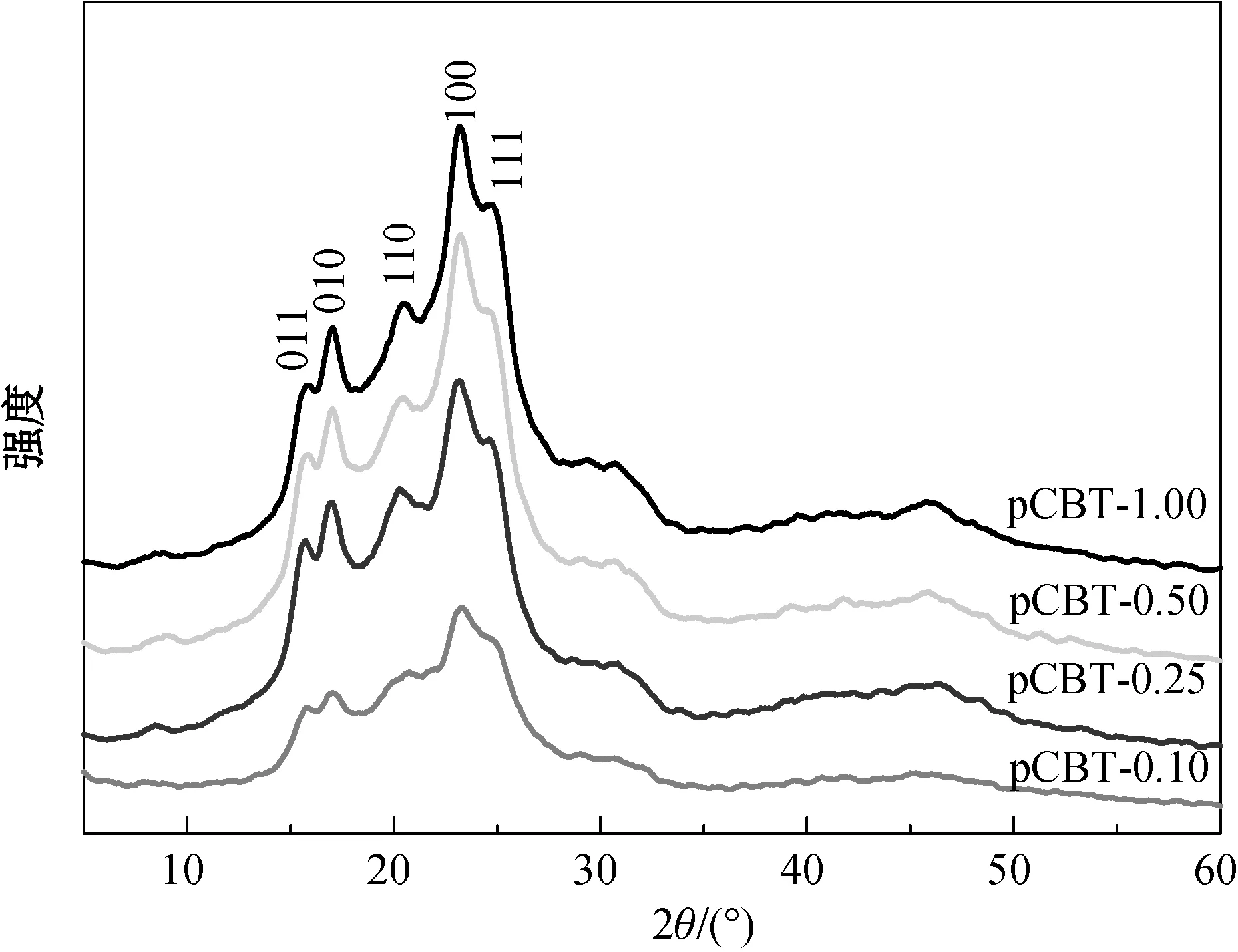
图5 不同引发剂用量条件下制得pCBT的XRD谱图Fig.5 XRD patterns of pCBT with different initiator content
3.5 pCBT的力学性能分析
不同引发剂用量条件下制得pCBT样条的力学性能如图6所示,具体数值列于表4中。由表4可知,引发剂用量对pCBT力学性能起着至关重要的作用。这是因为引发剂用量还会影响pCBT的结晶性能,进而影响材料的力学性能。由图6和表4可知,引发剂用量越大,pCBT分子量越小,结晶度越大,晶粒尺寸越小,材料的刚性越好,表现为其拉伸和弯曲模量越高。相反,分子量越大,结晶度越小,晶粒尺寸越大,材料韧性就越好,表现为其断裂伸长率和冲击强度越高。两者的综合作用使得材料的拉伸和弯曲强度随聚合引发剂用量的增大而先增大后降低。值得注意的是,引发剂质量分数低于0.5%时,pCBT制品在力学性能试验中均出现屈服现象,表现为典型的韧性断裂,而分子量较低的pCBT-1.0则没有出现屈服而直接发生断裂,变为明显的脆性材料,从而使其力学性能明显变差。
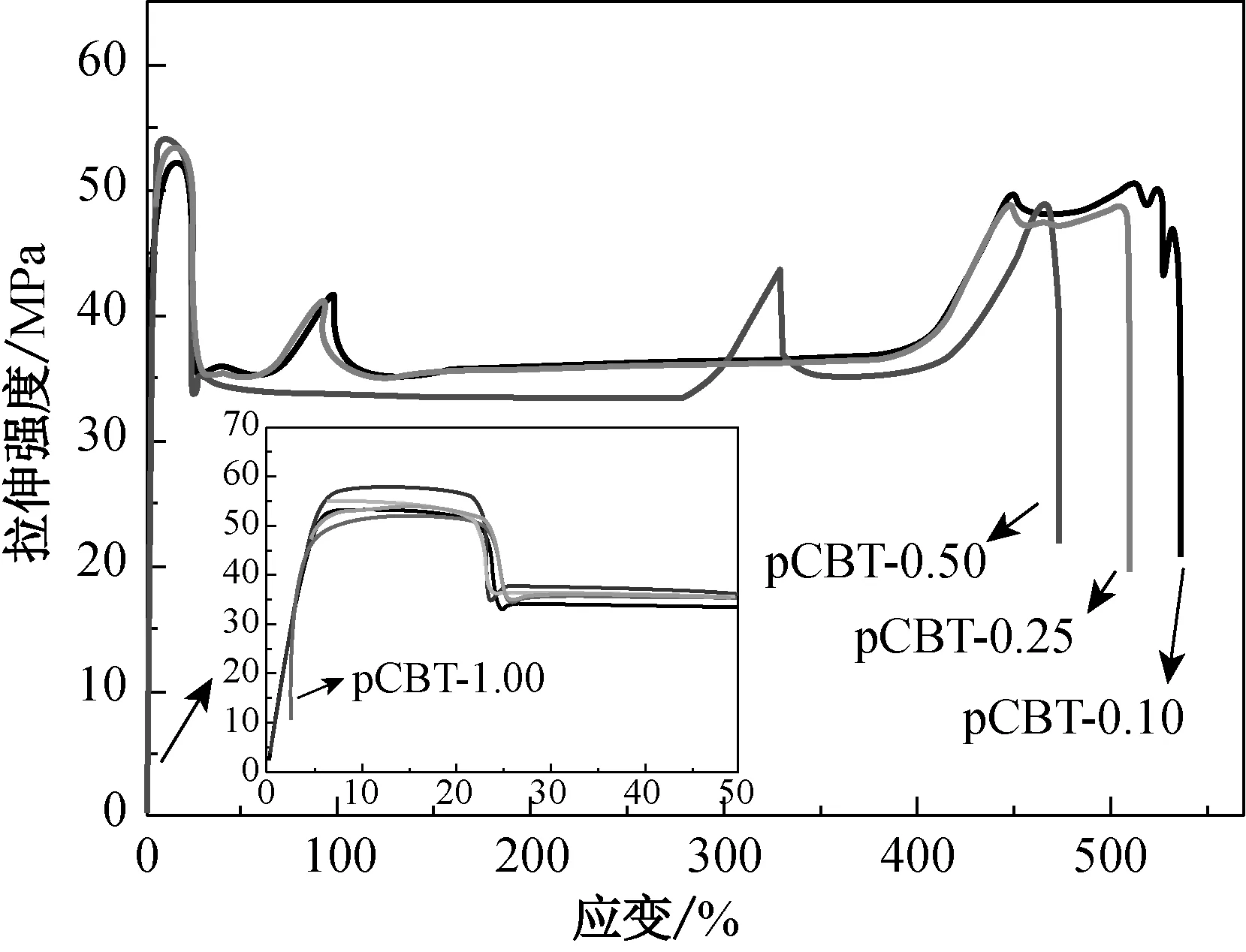
(a) 拉伸
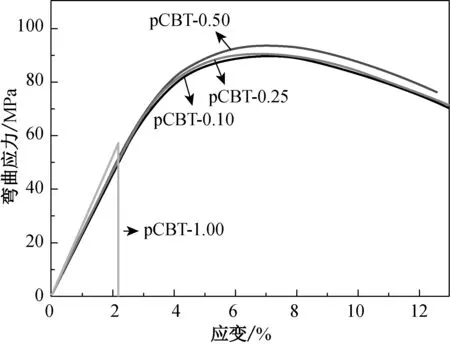
(b) 弯曲

样品拉伸强度/MPa拉伸模量/MPa断裂伸长率/%弯曲强度/MPa弯曲模量/MPa冲击强度/(kJ·m-2)pCBT-0.1052.3±0.31 113±6.7530±7.888.7±0.32 204±7.96.1±0.2pCBT-0.2553.6±0.21 205±3.2518±4.190.1±0.22 253±7.15.5±0.5pCBT-0.5054.3±0.11 254±2.9479±6.493.6±0.12 317±9.55.1±0.4pCBT-1.0028.0±0.61 312±5.2 4±2.640.0±0.92 540±8.51.4±0.3
4 结 论
(1) 本文使用平行板流变仪在线监测了不同引发剂含量的CBT在190 ℃条件下聚合时的流变过程,发现引发剂用量越多聚合反应开始得越早,且聚合速度越快,体系黏度上升得越迅速,低黏度区及凝胶时间越短,可用于加工的窗口越小。
(2) 对聚合产物的研究测试表明,随着引发剂用量的增大,制得pCBT分子量变小,结晶温度和最终熔融温度升高,结晶度变大,而晶粒尺寸则变小。
(3) 引发剂用量越大,pCBT拉伸和弯曲模量越高,其断裂伸长率和冲击强度越小,而拉伸和弯曲强度则先增大后减小。引发剂用量为0.5%时制得pCBT的综合力学性能最好,继续增大引发剂用量,材料的断裂方式由典型的韧性断裂转变为脆性断裂,力学性能明显下降。
