RHO1基因的HIGS表达载体构建及转基因水稻的抗病性分析
2018-08-10唐喆,张强,项萍
唐 喆,张 强,项 萍
(西北农林科技大学 植物保护学院,陕西 杨凌 712100)
真核生物中大量存在一些G蛋白,这些G蛋白在细胞分化和发育中起着重要的作用,其中Rho家族蛋白是G蛋白的重要组成部分[1-3]。已有研究发现,Rho家族蛋白在基因转录、囊泡运输、细胞运动和细胞增殖等方面具有调节作用,并且直接参与肌动蛋白在骨架形成时期的信号传导[4-5]。近年来,对真菌Rho家族蛋白结构和功能的研究也越来越多。研究报道,在酿酒酵母、芽殖酵母等真菌中RHO1基因及其同源基因与细胞极性生长、肌动蛋白重排和细胞壁形成以及细胞完整性直接相关[6-9]。在真菌粗糙脉孢霉和黑曲霉的rho1突变体研究中发现,RHO1基因对于这些丝状真菌细胞壁及细胞极性形成与维持具有重要作用[10-11]。研究发现,RHO1基因在稻瘟菌分生孢子萌发阶段表达量最高,对于稻瘟菌侵染水稻具有重要的作用[12]。
尚仁福等[13]研究发现,小分子RNA引起的RNA干扰会导致基因沉默,RNA干扰技术在促进植物与病原之间互作和提高植物抗病性方面有重要作用。近年来,利用RNA干扰技术发展起来的寄主诱导的基因沉默(HIGS)在植物中的研究迅猛发展[14-15]。HIGS是一种转录后的基因沉默技术[16]。当植物病原菌侵染含有HIGS载体的寄主时,转基因植株可产生特定序列的dsRNA,进而产生相应的小分子RNA,从而引起该病原菌关键基因表达量下降。病原菌内源的特定基因表达受到影响,从而使寄主获得对该病原菌的抗性。由此可见,该技术不仅可以从反向遗传学方向鉴定病原菌的遗传学特性,也能够通过沉默病原菌关键基因,提高植物抗病性。
稻瘟菌(Magnaportheoryzae)是引起水稻病害的一种病原真菌,它也是用于丝状真菌研究的重要模式生物之一。由稻瘟菌引起的稻瘟病是水稻的重要病害之一,能够引起水稻减产甚至绝收,严重影响了世界粮食安全。稻瘟病菌基因组序列已经被成功测序,分析结果表明,稻瘟菌可能存在11 109个基因[17]。将这些基因大致分为两类:一类是生长所需基因,它与菌体生长发育息息相关;一类是致病相关基因,在侵染寄主时具有重要作用。RHO1基因是生长所需基因,与其相互作用的蛋白涉及到病原菌生长发育繁殖等生物进程[18]。随着植物转基因技术的成熟,研究者已经将不同类型的外源基因转入到水稻中,如过量表达白藜芦醇合成酶基因(Resveratrol synthase,RS),能够提高转基因水稻对稻瘟病的耐性[19];过量表达病程相关蛋白(Pathogenesis-Related proteins,PRs)——β-1,3-葡聚糖酶(β-1,3-gulcanase),用于研究抗稻瘟菌水稻[20]。然而利用HIGS技术研究稻瘟菌和水稻之间相互作用的报道尚未见。为此,本研究选取了对稻瘟菌分生孢子萌发具有重要作用的RHO1基因,通过构建RHO1基因的HIGS载体,利用农杆菌介导转化进入日本晴水稻中,研究转基因水稻对稻瘟菌的抗性,为稻瘟病的综合防治提供理论依据。
1 材料与方法
1.1 供试菌株与试剂
稻瘟菌70-15、大肠杆菌DH5α、农杆菌AGL1、质粒 pDONR 221(Kan+)、水稻日本晴种子,均为西农林科技大学许金荣实验室保存;质粒pBDL03,中国农业科学院惠赠;大肠杆菌DH5α和农杆菌AGL1感受态细胞由许金荣实验室制备;TRIzol®Reagent、Gateway®BP ClonaseTMⅡ Enzyme Mix、Gateway®LR ClonaseTMⅡ Enzyme Mix,均购于Invitrogen公司;快速内切酶(KpnⅠ、SacⅠ、EcoR Ⅴ),dNTP,TaqDNA聚合酶,均购于Fermentas公司;抗生素(卡那霉素(Kan)、潮霉素(Hyg)、利福平(Rif))购于Roche公司;凝胶回收试剂盒、质粒小量提取试剂盒,均购于百泰克公司;其他试剂为国产化学纯,引物合成和测序由北京奥科公司完成。
1.2 RNA干扰序列的选择与引物设计
通过查询稻瘟菌基因组数据库(http://fungi.ensembl.org/Magnaporthe_oryzae/Info/Index),获得RHO1基因(MGG_07176)的编码序列(CDS)区域。利用BLOCK-iTTMRNAi Designer在线工具(https://rnaidesigner.thermofisher.com/rnaiexpress/design.do)查找适合用于沉默的片段。利用 Primer 5.0设计干扰区特异性引物RHO1-F和RHO1-R。该片段不含EcoRⅤ、SacⅠ、KpnⅠ的酶切位点。引物序列如下:RHO1-F.5′-GGGGA-CAAGTTTGTACAAAAAAGCAGGCTTCCGTC-GCTGATGTGGAGGT-3′,RHO1-R.5′-GGGGACCACTTTGTACAAGAAAGCT-GGGTC CGAACA-CCTCACGGACACC-3′,其中引物序列中添加了Gateway的接头序列(下划线所示)。
1.3 RNA提取与目标干扰片段的获得
利用TRIzol(按照试剂操作说明)提取稻瘟菌70-15的总RNA。利用反转录试剂盒进行RNA反转录,获得cDNA。然后以该cDNA为模板,利用PCR技术进行特异性干扰片段的扩增。PCR扩增体系(50 μL): cDNA 1 μL,10 μmol/L的特异性引物RHO1-F和RHO1-R各2 μL,10×PCR buffer(含MgCl2) 5 μL,dNTPs 2 μL,TaqDNA聚合酶0.6 μL,ddH2O 36.4 μL。PCR扩增程序: 94 ℃ 预变性 5 min;94 ℃ 30 s,58 ℃ 40 s,72 ℃ 30 s,35个循环;72 ℃后延伸10 min, 16 ℃保存。PCR产物利用1.5%琼脂糖凝胶电泳进行检测,验证其条带长度正确后,使用凝胶回收试剂盒回收目标条带,并测定其浓度。
1.4 HIGS表达载体的构建
1.4.1 入门载体构建 利用Gateway试剂盒进行BP反应,构建入门载体 pDONR 221-RHO1(图1)。
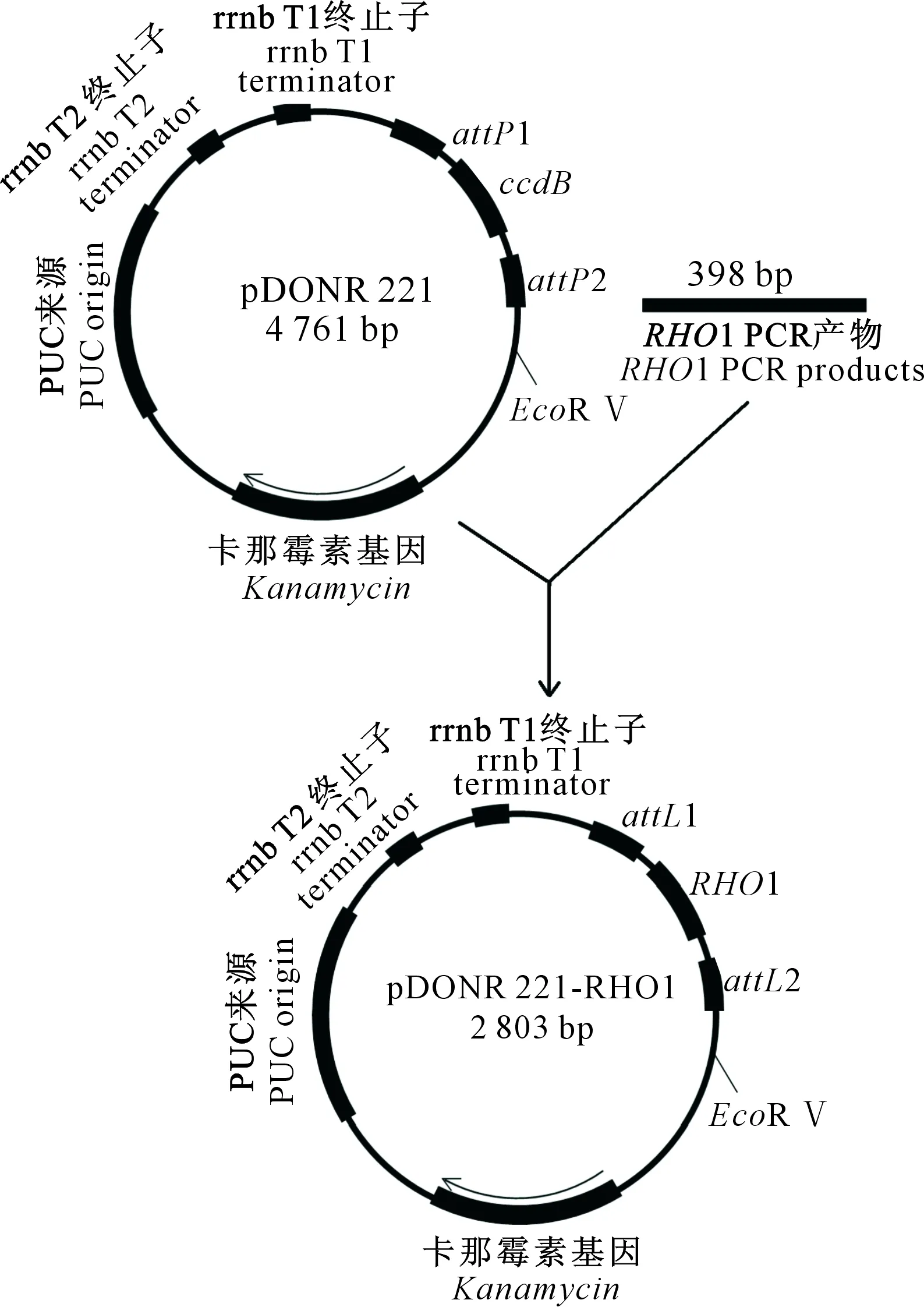
图1 基于pDONR 221载体与稻瘟菌RHO1基因片段构建入门载体pDONR 221-RHO1示意图Fig.1 Construction of pDONR 221-RHO1 by recombination of pDONR 221 plasmid and rice blast RHO1 gene fragment
用凝胶电泳回收纯化后具有接头序列的PCR产物与载体 pDONR 221进行基因重组反应。按照试剂使用说明,采用下列体系:PCR 产物 2 μL(100 ng),质粒 pDONR 221 1 μL(150 ng),BP克隆混合酶溶液1 μL,TE缓冲液(pH 8.0)补至5 μL。轻轻混合均匀,25 ℃孵育过夜;加入1 μL蛋白酶K溶液(100 μmol/L)混匀,37 ℃孵育10 min;终止BP反应。将BP反应后得到的产物稀释5倍,取2 μL反应产物进行电击转化,转入大肠杆菌DH5α感受态细胞,转化后涂布在含50 mg/L卡那霉素的LB筛选平板上,37 ℃培养过夜。挑取筛选平板上单克隆的大肠杆菌DH5α菌落,在10 mL含有50 mg/L卡那霉素的LB溶液中培养过夜,然后提取质粒pDONR 221-RHO1,利用RHO1-F和RHO1-R 进行PCR检测,以获得阳性克隆。将阳性克隆送奥科公司基因测序,测序正确即成功构建入门载体。
1.4.2 表达载体构建 将测序正确的入门载体pDONR 221-RHO1,利用EcoRⅤ内切酶进行线性化,反应体系如下:质粒pDONR 221-RHO1 2 μg,EcoRⅤ 1 μL,buffer 2 μL,ddH2O补至20 μL,37 ℃孵育20 min, 80 ℃处理5 min,酶切产物利用1.5%琼脂糖凝胶电泳进行检测,使用凝胶回收试剂盒回收目标条带,并测定其浓度。
线性化的入门载体 pDONR 221-RHO1在RNAi干扰片段两端具有attL1和attL2 2个重组位点,表达载体pBDL03含有attR1和attR2 2个重组位点,二者在Gateway LR反应中能够发生同源重组,表达载体上的attR1和attR2之间的序列ccdB被线性化入门载体上RHO1的RNAi干扰片段替换,从而构建成完整的HIGS表达载体pBDL03-RHO1(图2)。
Gateway LR反应构建HIGS表达载体,按照试剂盒使用说明,采取下列反应体系:线性化入门载体pDONR 221-RHO1 100 ng,pBDL03 空载体 100 ng,Gateway LR反应酶混合物(LR enzyme mix) 1 μL,TE缓冲液(pH 8.0)补至5 μL,轻轻混匀,25 ℃孵育过夜。加入1 μL 蛋白酶K溶液(100 μmol/L)混匀,37 ℃孵育10 min,终止Gateway LR反应。将Gateway LR反应产物稀释5倍,取2 μL反应产物进行电击转化,转入大肠杆菌DH5α感受态细胞,转化后涂布在含50 mg/L卡那霉素的LB筛选平板上,37 ℃过夜培养。挑取筛选平板上单克隆的大肠杆菌 DH5α菌落,在10 mL含有50 mg/L卡那霉素的LB溶液中过夜培养,然后提取质粒pBDL03-RHO1,利用SacⅠ和KpnⅠ双酶切进行鉴定。利用pBDL03空载体作为对照,鉴定阳性克隆。利用在GUS linker处设计的测序引物(GUSLR1:5′-GACCA AAGCC AGTA AAGTA GAACG-3′,GUSLF2:5′-CGTCG GTGAA CAGGT ATGGA-A-3′)测定靶标序列。测序正确表明成功构建了HIGS表达载体pBDL03-RHO1。

图2 HIGS表达载体pBDL03-RHO1构建示意图Fig.2 Construction of HIGS expression vector pBDL03-RHO1
1.5 转基因植物的获得与鉴定
利用农杆菌电转化法将构建的HIGS载体pBDL03-RHO1 转化进入农杆菌AGL1感受态细胞,转化后涂布在含50 mg/L卡那霉素和50 mg/L利福平的LB筛选平板上,28 ℃培养36~48 h,从筛选平板上选取单克隆农杆菌,液体摇培,然后提取质粒,利用SacⅠ和KpnⅠ双酶切进行鉴定,鉴定正确的农杆菌可用于水稻转化。
参照Hiei等[21]的方法进行水稻日本晴的转化。将水稻日本晴的成熟愈伤组织和携有HIGS载体pBDL03-RHO1的农杆菌AGL1进行共培养,经过含有潮霉素的培养基进行2~3次筛选,得到含有抗性的愈伤组织,经过分化再生,得到正确的水稻日本晴的转基因株系。
利用HIGS载体pBDL03-RHO1上带有的红色荧光蛋白标记基因mCherry,观察红色荧光[22-23];利用潮霉素基因以及RHO1基因的RNA干扰片段,进行PCR扩增检测,以获得阳性转基因植株。
1.6 转基因植株的抗病性分析
对于鉴定正确的转基因植株,从中随机选取5株进行稻瘟菌孢子接种试验[24]。收集成熟的稻瘟菌分生孢子,用0.25%明胶溶液将稻瘟菌分生孢子配制成1×105mL-1孢子悬浮液,用喷雾器将该孢子悬浮液均匀地喷到3叶期的水稻日本晴(包括野生型和转基因植株)叶片上,25 ℃黑暗条件下保湿2 d,然后进行正常培养,接种7 d后对水稻叶片上病斑进行观察,选取接种水稻叶片中部(长度约为5 cm)进行病斑计数。以野生型水稻叶片上病斑数量为1,计算转基因水稻相对病斑数,进而分析其抗病性。
2 结果与分析
2.1 RHO1基因片段的获得
通过查询稻瘟菌基因组数据库得到RHO1的CDS序列,利用BLOCK-iTTMRNAi Designer工具分析设计最佳的RNA干扰片段,以该片段序列设计获得特异性引物RHO1-F和RHO1-R。提取稻瘟菌70-15的总RNA,经过反转录得到的cDNA为模板,用引物RHO1-F和RHO1-R扩增获得特异性片段,其长度为389 bp (图3)。
2.2 BP反应构建入门载体 pDONR 221-RHO1及其鉴定
将RHO1基因片段PCR产物连接到入门载体pDONR 221后,随机选择几个单克隆的菌落,摇培后提取质粒。以质粒为模板,RHO1-F和RHO1-R为引物进行PCR检测,结果显示片段长度为400 bp左右,将其送奥科公司对pDONR 221-RHO1测序,测序比对结果(图4)表明,RHO1基因片段已成功转入入门载体pDONR 221。
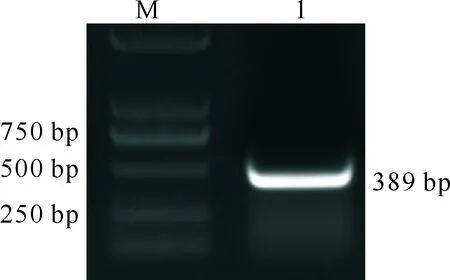
M.DNA Marker DS 2000;1.扩增的RHO1基因M.DNA Marker DS 2000;1.Amplification of RHO1 gene 图3 稻瘟菌基因RHO1片段检测结果Fig.3 Detection of rice blast gene RHO1 fragment

*所示为构建pDONR 221-RHO1测序结果中与RHO1基因片段完全比对上的碱基序列* means the same sequence of vector pDONR 221-RHO1 alignment with RHO1 gene图4 构建的质粒pDONR 221-RHO1与RHO1基因序列的比对Fig.4 Sequencing of vector pDONR 221-RHO1 alignment with RHO1 gene
2.3 LR反应构建HIGS表达载体pBDL03-RHO1及其鉴定
图5显示,构建正确的重组HIGS表达载体pBDL03-RHO1经SacⅠ和KpnⅠ双酶切后,通过电泳分离可分别获得长度约为10和2 kb的条带,与预期结果一致,表明重组质粒构建成功。
2.4 转基因植物的获得与鉴定
将成功构建的HIGS表达载体pBDL03-RHO1电转化进入农杆菌AGL1感受态细胞,经过筛选获得含有HIGS载体的农杆菌菌株。采用农杆菌介导的转基因方法将HIGS表达载体转化进入水稻中,获得含有HIGS表达载体的转基因水稻植株。经过培养后收获种子,通过繁殖获得F1代植株,通过mCherry红色荧光观察初步鉴定其为阳性转基因植株。
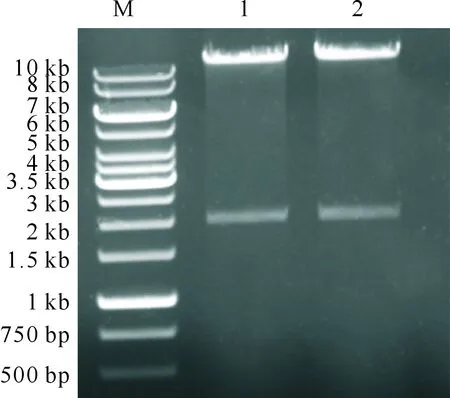
M.DNA Marker DS 2000;1~2.阳性克隆质粒酶切结果M.DNA Marker DS 2000;1-2.Digested plasmid of positive clone图5 表达载体pBDL03-RHO1的SacⅠ和KpnⅠ双酶切鉴定结果Fig.5 Verification of expression vector pBDL03-RHO1 by SacⅠ and KpnⅠ double restriction enzyme digestion
提取F1代转基因水稻的总DNA,使用潮霉素基因片段的特异性引物和转入的RHO1基因片段的特异性引物进行鉴定。PCR扩增结果(图6和图7)显示,野生型水稻日本晴中不存在潮霉素基因片段和RHO1基因片段,而在阳性的转基因水稻中扩增到了特异的潮霉素基因片段(750 bp)和RHO1基因片段(389 bp),与预期结果一致,表明含有RHO1基因的HIGS表达载体的转基因水稻植株构建成功。
2.5 HIGS转基因植株的抗病性
将鉴定的F1代阳性转基因植株培养至3叶期时,接种稻瘟菌70-15,分析转基因植株抗病性。图8显示,与野生型水稻日本晴相比,HIGS转基因植株对稻瘟菌的抗性提高,但不同转基因株系之间抗病性略有差异。统计分析表明,与野生型相比,HIGS转基因植株叶片上的病斑数时显减少(图9)。这表明HIGS转基因植株对稻瘟菌具有抗性,但不同株系之间抗病性存在一定的差异。

M.DNA Marker DS 2000;1~5.转化pBDL03-RHO1的阳性植株;WT.野生型水稻日本晴M.DNA Marker DS 2000;1-5.Transgenic plants carrying pBDL03-RHO1;WT.Wild type rice图6 转基因水稻潮霉素片段PCR检测结果Fig.6 PCR detection of HYG fragment in transgenic rice plants

WT.野生型水稻日本晴;1~5.转基因水稻 WT.Wild type rice;1-5.Transgenic rice图8 稻瘟菌70-15在野生型和HIGS转基因水稻上的发病情况Fig.8 Disease symptom developed on the wild type and transgenic rice inoculated by Magnaporthe oryzae strain 70-15
3 讨 论
传统构建表达载体的方法是通过酶切连接的方法将目的基因连接入表达载体,酶切位点的选择、合适的表达载体选择以及连接进入载体正确方向的保证都是传统表达载体构建的难点和重点。有时由于缺乏合适的酶切位点,则需采用平端位点连接,更是增加了筛选阳性克隆的工作量。本研究利用Gateway技术的特异位点重组技术,构建了稻瘟菌RHO1基因的HIGS表达载体,这种技术具有简单方便、转化效率高的特点。该方法只需要两步就可以将外源基因导入到HIGS表达载体。第一步,经过Gateway的BP反应,将外源基因RHO1连接到入门载体pDONR 221上;第二步,利用Gateway的LR反应,将pDONR 221上的RHO1片段整合到pBDL03中,得到HIGS表达载体pBDL03-RHO1。最终构建的载体由UBi启动子驱动,GUS linker 连接的两个反向序列产生发卡结构,形成dsRNA,可以进行下一步干扰试验。
水稻转基因试验具有周期长、工作量大等特点,快速准确鉴定阳性植株,区分假阳性和阴性对照,是保证试验顺利进行的关键技术。本研究将构建好的pBDL03-RHO1表达载体,利用农杆菌AGL1,通过农杆菌介导的方法转化进入水稻日本晴中,获得了转基因植株。选取的pBDL03-RHO1表达载体有众多的筛选标记,阳性转基因植株可通过观察mCherry红色荧光蛋白、检测潮霉素基因和检测转入的RNAi基因RHO1片段来鉴定。试验结果表明,3种检测手段结果一致,均可用于转基因植株的检测。初步检测利用观察mCherry红色荧光蛋白,简单、方便、成本较低,为大量筛选阳性的转基因植株节省了时间、人力、物力和财力,可以大大提高阳性转基因植株的筛选效率。进一步检测转入的载体上的抗性基因和转入的外源干扰基因片段,保证了初筛的阳性转基因植株的结果更加准确和可靠。
RNA干扰技术具有特异高效、操作简单方便等特点,近年来被广泛应用到基因功能的研究中。随着RNA干扰技术的发展,寄主诱导的基因沉默(HIGS)在植物中的应用研究快速发展。本研究成功获得了含有RHO1基因的水稻HIGS转基因植株,该转基因植株含有稻瘟菌70-15的RHO1基因为靶标的dsRNA,当稻瘟菌侵染该水稻时,该dsRNA具有引发RNAi干扰的潜力,沉默RHO1基因,影响RHO1的表达,从而影响稻瘟菌对植物的侵染。接种试验结果表明,与野生型水稻相比,转基因水稻病斑数减少,表明稻瘟菌对该转基因水稻的侵染力有一定程度的降低。然而,相比于HIGS在作物抗布氏白粉菌[25]和轮枝镰孢菌[26]侵染中的应用,本试验中转基因水稻的抗病性还有待进一步优化提高。
近年来,HIGS技术迅猛发展,已经成为控制真菌病害的重要手段之一[25-26],其在抗病转基因作物的培育方面具有广阔的应用前景。该技术通过将病原菌关键基因的同源序列转入植株,并在植物体内形成dsRNA,以达到干扰病原菌关键基因表达的目的,从而降低病原菌定殖水平,提高植物抗病性。与此同时,该方法针对特定的病原菌具有更高的特异性,为培育遗传稳定、特异性强和环境安全的转基因作物奠定了基础。然而,并不是所有的HIGS转基因作物均可沉默病原菌基因并达到预期效果[27]。如何有效确定关键的用于沉默的基因片段,怎样构建方便使用的基因表达载体等问题,对于不同真菌和植物互作研究以及转基因植物的抗病性获得非常必要,因此该技术还有待进一步发展和完善。
