ZnO量子点与尿酸分子界面特性的理论研究
2018-08-06郭纪源陈平超苗德壮吴升聪刘升卫
郭纪源,陈平超,苗德壮,吴升聪,刘升卫
(江苏科技大学 理学院,镇江 212003)
纳米材料与生物材料的复合体系的特性使得它们在纳米电子学、信息技术、生物医学工程和界面科学等多个重要领域得到了应用[1-3].而基于这些复合材料体系制备的纳米生物传感器,由于综合光、声、电、色等多种先进检测技术,对环境检测、疾病诊断、生物反恐、遗传分析等领域产生革命性影响,已经成为国际上研究的前沿和热点[1-6].早期的生物传感器是以氧作为酶与电极之间的电子通道;之后,人们使用小分子作为电子媒介替代氧;而今,基于纳米材料量子尺寸效应和表面效应的第3代生物传感器利用检测物自身与电极之间的直接相互作用来完成信号转换.因此,开展被检测生物分子与电极介质之间的直接界面特性研究十分必要.
氧化锌作为宽禁带半导体材料,具有制备容易、纳米结构形貌丰富、光学性质优异、电子传导能力良好、无毒、生物兼容性优良、生物安全和环境友好等优点[4-7],其高达9.5的等电点,使得ZnO纳米结构表面的生物分子组装和修饰更易操作且可靠[5].目前,氧化锌已经在生物领域展开应用,包括生物传感、纺织、表面喷涂、药物合成、护肤品、口腔医学等,特别是用于伤口部位,抑制大肠杆菌、金黄色葡萄球菌生长,或被作为试验药物来治疗中枢神经系统疾病[8-12]等.尿酸是嘌呤代谢的最终产物,其浓度异常是人体某些疾病的反映,例如痛风仍困扰着众多的患者.因此,研制一种准确、简单的测量尿酸含量的传感器有着重要的意义.常用的尿酸检测方法有酶分析法、伏安法、高效液相色谱法等. 酶分析法、高效液相色谱法灵敏度高,但所需试剂和仪器价格昂贵,难普及.现有的报道中,人们已经采用ZnO纳米棒、微米线、薄膜、四针状纳材料[13-16]等,通过组装尿酸酶来作为尿酸的检测器,实验结果都显示其有较好的检测效果和较好的稳定性.直接基于ZnO纳米材料与尿酸相互作用的传感器未见报道,原因是上述ZnO纳米材料与尿酸相互作用的转换信号极其微弱.
文中采用SCC-DFTB(self-consistent charge density functional tight-binding)与DFT(density functional theory)结合的方法,以(ZnO)12团簇作为ZnO量子点模型,对ZnO量子点与尿酸分子的界面特性进行了理论研究.在得到该复合体系的最优化结合体系之后,通过分析结构之间成键、结合能和电荷转移等,判断ZnO量子点与尿酸分子之间的相互作用强弱,为设计ZnO量子点直接检测尿酸分子的传感器提供参考.
1 模型与方法
文中采用基于电荷自洽的密度泛函-紧束缚(SCC-DFTB)方法[17]的DFTB软件包[18]进行结构优化计算,该软件包在德国Marcus Elstner课题组的领导下开发,主要为了适应生物和材料领域大规模分子动力学反应,固体电子结构及其对外界扰动的响应和输运性质计算的需求.DFTB方法对分子轨道进行了双中心近似处理,可以视为使用最小基的、近似的密度泛函方法.SCC-DFTB方法是Kohn-Sham方程总能量对电荷密度变化进行泰勒级数二级展开.它对交换积分进行近似和参数化,紧束缚近似简化后的哈密顿矩阵元由电荷自洽决定.分子轨道自洽要计算不同原子对间的原子轨道积分,但因为原子轨道是冻结的,它们的积分可以预先计算,然后保存到参数文件中.文中Zn与X(X=H,C,N,O和Zn)元素之间的参数化Slater-Koster 文件包含在压缩包znorg-0-1里,C,H,O,N之间的参数化Slater-Koster 文件包含在压缩包mio-1-1里面,这两个压缩包中的参数文件都是经过测试且公开发表的文件[19-20],可以在DFTB官方网站上下载,计算中采用了Universal Force Field 参数[21],用Lennard-Jones方法[22]进行了色散力修正.与VASP、Quantum Espresso和Siesta等代表的DFT计算软件相比,尽管DFTB能量计算精度稍差,但是采用测试后的参数化Slater-Koster 文件,结构计算所得到的键长键角等参数较好;而对于DFT低估体系的带隙问题,DFTB计算结果更能接近实验值,更有利于研究由于吸附、缺陷等引起的电子结构的变化[20].此外,DFTB由于方法简化,比DFT具有更快的计算速度.
(ZnO)12团簇的量子点模型如图1(a),由6个(ZnO)2四元环和8个(ZnO)3六元环组成,是一个Zn原子和O原子对称分布的笼状结构.该团簇的初始模型坐标参考文献[23].尿酸分子坐标参考文献[24].结构弛豫使用共扼梯度算法优化了所有原子位置,原子受力的收敛精度为5×10-4eV/Å,电子自洽计算的能量收敛判据为1×10-6.在DFTB输入文件中,所有构型模式均设置为非周期性的团簇模式.由于DFTB里面采用Mulliken电荷分析,精度较差,为了获得较为准确的电荷分布信息,使用QE(Quantum-Espresso)软件[25],采用公认的更为准确的Bader电荷分析方法,对(ZnO)12与尿酸分子的最稳定结构进行差分电荷密度计算,QE计算时采用模守恒贋势,交换关联势采用广义梯度近似,波函数截断能为60 Ry,电荷密度截断能为240 Ry,原子力的收敛精度为10-6eV/Å.

图1 优化后的(ZnO)12量子点和尿酸分子构型Fig.1 Optimized configuration of (ZnO)12 quantum dot and uric acid molecule
2 结果与讨论
首先进行程序的验证性计算,单独计算(ZnO)12量子点,把计算结果与其他文献结果进行对比.经过优化后的构型图见图1(a).表1列出了计算数据与其他文献中数据的对比,主要从键长、键角和能隙3个方面进行比较.根据图1(a)和表1,SCC-DFTB计算得到的键长与文献[26-28]中数据的对比误差范围为1.5%~2.0%;键角与文献[26-28]中数据的对比误差范围为0.4%~5.5%;能隙值与文献[26-27]中结果的误差为0.7%;与
文献[28-29]中软件DFT计算的值相差比较大,(ZnO)12团簇能隙的实验值未见报道.为了进一步验证准确性,用SCC-DFTB计算了ZnO块体周期性结构的能带(图2),得到带隙为3.78 eV,与文献[30]中采用同样计算方法得到的结果完全一致,而实验报道的值为3.37 eV.由于量子效应,量子点的能隙比其自身块体材料的大些.从这些数据来看,运行的SCC-DFTB程序是成功的.
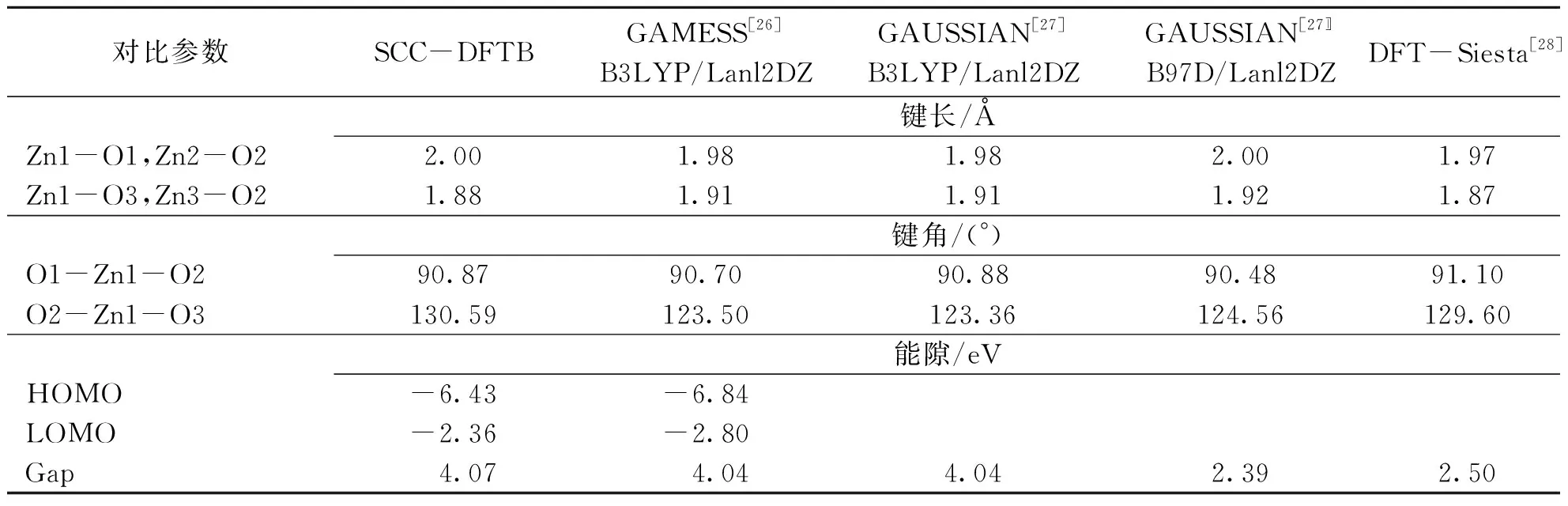
表1 (ZnO)12量子点结构优化后的各项数据对比Table 1 Calculated results of (ZnO)12 quantum dot and its comparision with other reported results

图2SCC-DFTB方法计算得到的ZnO
块体材料的能带结构
Fig.2BandstructureofbulkZnOwhichwascalculated
bytheSCC-DFTBmethod
尿酸分子是一个平面分子(图1(b)),采用SCC-DFTB进行结构优化计算后得到的构型、键长、键角和二面角数据如表2,与文献[31]中的数据相比,键长误差范围为0.5%~3.6%,键角度误差范围为0.2%~4.9%.而二面角的数据则进一步说明了该分子的平面特性.图3为尿酸分子的各种构型方位.

图3 尿酸分子构型的不同方位Fig.3 Different binding sites of uric acid molecule表2 尿酸分子优化后的部分键长和键角值Table 2 Optimized bond lengths and bond angles of the uric acid molecule
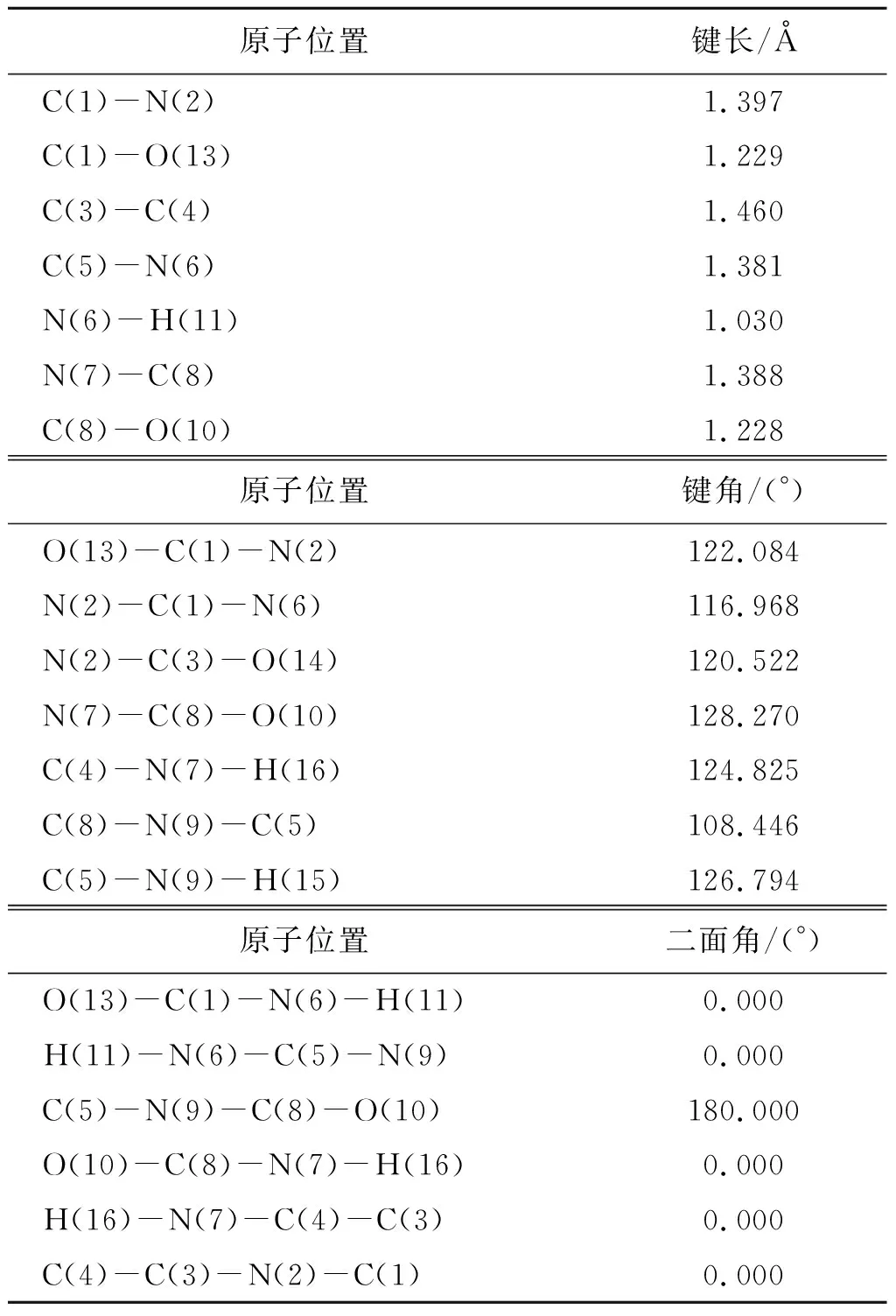
原子位置键长/ÅC(1)-N(2)1.397C(1)-O(13) 1.229C(3)-C(4) 1.460C(5)-N(6) 1.381N(6)-H(11)1.030N(7)-C(8)1.388C(8)-O(10)1.228原子位置键角/(°)O(13)-C(1)-N(2)122.084N(2)-C(1)-N(6)116.968N(2)-C(3)-O(14) 120.522N(7)-C(8)-O(10)128.270C(4)-N(7)-H(16)124.825C(8)-N(9)-C(5)108.446C(5)-N(9)-H(15)126.794原子位置二面角/(°)O(13)-C(1)-N(6)-H(11)0.000H(11)-N(6)-C(5)-N(9)0.000C(5)-N(9)-C(8)-O(10)180.000O(10)-C(8)-N(7)-H(16)0.000H(16)-N(7)-C(4)-C(3)0.000C(4)-C(3)-N(2)-C(1)0.000



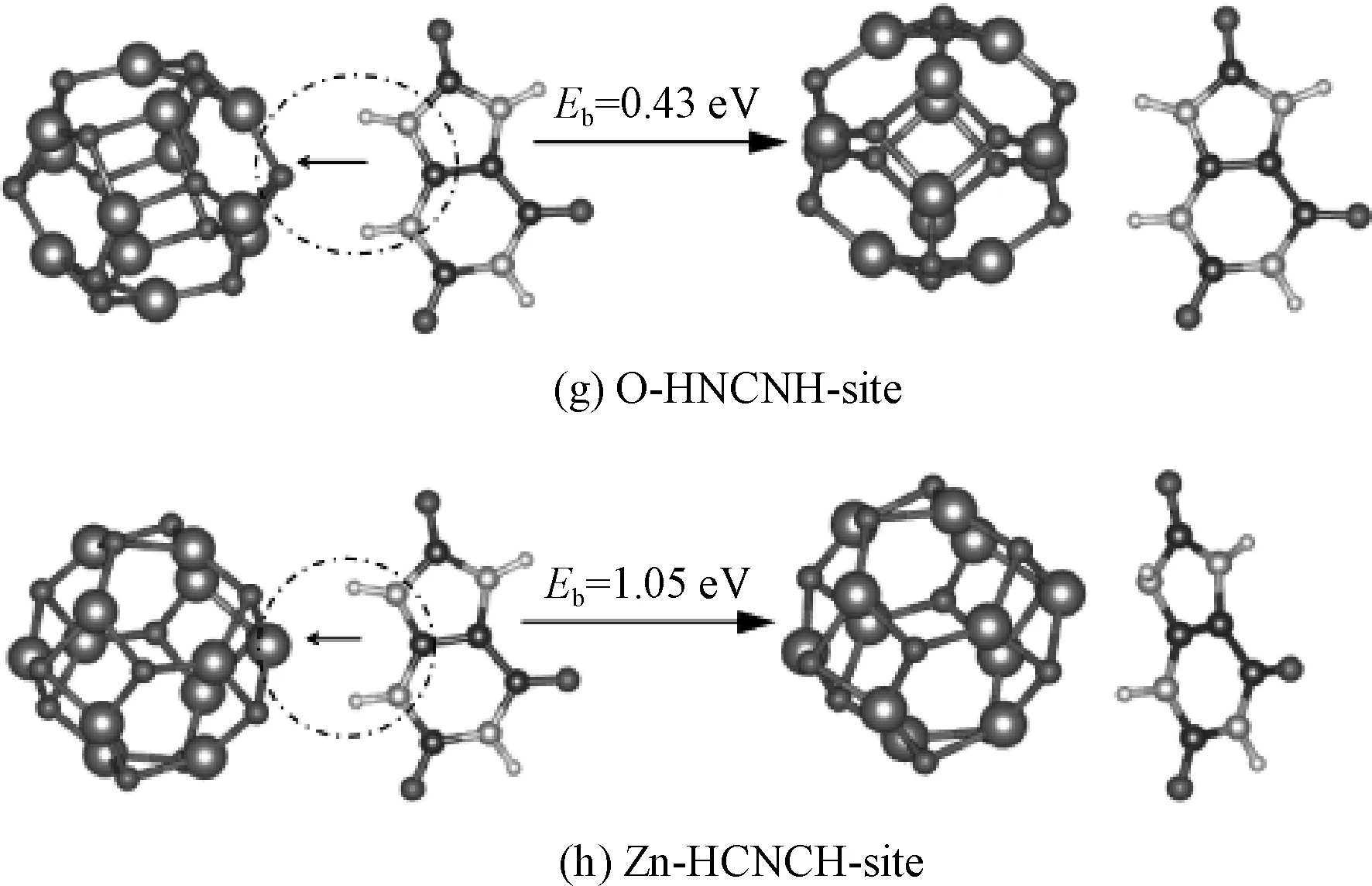
图4 初始构型与优化构型Fig.4 Preliminary stractures and converged caufiguration
按照文献[28]中的经验,尿酸分子的这些不同方位,与(ZnO)12量子点构建了不同的初始构型.以尿酸分子的不同方位靠近(ZnO)12量子点的Zn位置或O位置.典型的初始构型如图4的左边.图中,虚线圈代表ZnO量子点与尿酸分子的相对位置,初始相对位置中,两者最短距离为3 Å.复合体系结合能的计算方式采用文献[28]中的方法,(ZnO)12与尿酸分子相距10 Å时的总能量减去复合体系不同构型时最后优化后的构型总能量,这个差值就定义为结合能.每种不同构型最后优化后的构型总能量,被认为是尿酸分子以不同方位接近(ZnO)12后得到的最低能量.图4中的右边为优化后的收敛构型,d为成键距离,Eb为结合能.
从各种初始模型优化的最后结果来看,(ZnO)12量子点和尿酸分子基本都保持了各自原来的构型,这说明两者都具有比较好的稳定性.对(ZnO)12量子点而言,可以以Zn原子去结合尿酸分子,也可以以O原子去结合尿酸分子.计算结果显示,以Zn原子去结合尿酸分子,总能得到收敛的复合构型,而以O原子去结合尿酸分子,优化过程中,还会被调整为以Zn原子与尿酸分子进行结合,如图4 (e).这说明以O原子去结合尿酸分子得不到稳定构型,文献[28]在研究(ZnO)12与核酸碱基的不同方位构型时,也发现了这个特性.在图4中,尿酸分子以不同的方位去靠近(ZnO)12量子点,优化收敛过程都是调整尿酸分子中最近的O原子去靠近量子点,最后与量子点上Zn原子成键.而如果尿酸分子中的所有O原子与(ZnO)12量子点距离都足够远,最后的收敛构型中,尿酸分子与(ZnO)12量子点之间没有成键,如图4(g)和4(h).
对比所有构型中成键的距离值和结合能数据,键长越短结合能越小.(ZnO)12量子点上Zn原子与尿酸分子上的O原子的成键距离在2.08~2.23 Å之间,接近(ZnO)12量子点中Zn原子与O原子的距离(1.88~2.00 Å).而图4(e)中,尿酸分子有两个O原子与(ZnO)12量子点上两个Zn原子成键,这个构型是测试的所有构型中结合能最大的构型,达到了1.59 eV,结合能较大,说明这个复合体系一旦形成,之后是比较稳定的.这个结合过程会释放比较多的能量,这个能量越大,对于观察(ZnO)12量子点与尿酸分子的反应越有利;同时,也意味着反应活性强,容易检查到反应信号.按照能量最低原理,在(ZnO)12量子点与尿酸分子界面反应中,以这个构型结合的概率最大.采用图4(e)得到的优化结构作为QE软件的初始构型.从构型数据上分析,DFT优化计算得到的构型与SCC-DFTB计算得到的构型几乎没有变化.接着进行自洽计算,采用Bader电荷分析方法,得到了(ZnO)12量子点和尿酸分子的电荷密度分布图,以及它们复合体系的差分电荷密度图(图5).图5(a)中,电荷聚集在O原子上,而Zn原子都失去电子,处于电荷耗散区.图5(b)中,电荷主要聚集在尿酸分子的O原子、4个N原子和1个C原子上.图5(c)中,尿酸分子与量子点成键的Zn原子上出现了电荷聚集,尿酸分子与(ZnO)12量子点成键的两个O原子上的电荷往量子点成键的Zn原子转移了,如虚线圈所示.根据计算数据分析,转移电荷量约为0.344 e.

图5 电荷密度及复合个体系的差密度Fig.5 Charge densities and charge density difference of the complicated system
综合上述界面特性计算结果,尿酸分子中的O原子倾向于与(ZnO)12量子点上的Zn原子成键,两者间存在较强的相互作用,界面之间也存在电荷转移.根据这些界面特性的数据结果可知,(ZnO)12量子点与尿酸的作用过程中,会有较多的能量释放,这为使用(ZnO)12量子点直接检检测一定浓度的尿酸分子提供了可能.初步的实验方案为先制备比较分散的ZnO量子点,在溶液中放入电化学发光材料,再加入尿酸,尿酸分子与ZnO量子点的反应释放出能量,这个能量被发光材料吸收后发光,就可以通过统计材料发光的强弱来判断尿酸的浓度;此外,从电荷转移结果出发,也可以通过观察尿酸分子与ZnO量子点反应引起的电信号变化来实现检测.
3 结语
文中采用SCC-DFTB与DFT理论计算方法,对ZnO量子点与尿酸分子的界面特性进行了模拟研究,结果显示尿酸分子上的O原子与ZnO量子点上的Zn原子有较强的成键相互作用,最优化结构的结合能达到1.59 eV,通过Bader电荷分析,ZnO量子点与尿酸分子界面间存在明显电荷转移.基于计算结果,下一步一方面拟开展ZnO量子点直接检测尿酸分子传感器的实验制备工作;另一方面,基于SCC-DFTB能隙计算优势,将在ZnO量子点掺杂及缺陷等情况下,开展与生物分子界面特性相互作用的研究.
