Sequencing, de novo assembly and characterization of the spotted scat Scatophagus argus (Linnaeus 1766) transcriptome for discovery of reproduction related genes and SSRs*
2018-08-02YANGWei杨尉CHENHuapu陈华谱CUIXuefan崔雪凡ZHANGKewei张克伟JIANGDongneng江东能DENGSiping邓思平ZHUChunhua朱春华LIGuangli李广丽
YANG Wei (杨尉) CHEN Huapu (陈华谱) CUI Xuefan (崔雪凡) ZHANG Kewei (张克伟) JIANG Dongneng (江东能) DENG Siping (邓思平) ZHU Chunhua (朱春华) LI Guangli (李广丽)
Abstract Spotted scat ( Scatophagus argus) is an economically important farmed fish, particularly in East and Southeast Asia. Because there has been little research on reproductive development and regulation in this species, the lack of a mature arti ficial reproduction technology remains a barrier for the sustainable development of the aquaculture industry. More genetic and genomic background knowledge is urgently needed for an in-depth understanding of the molecular mechanism of reproductive process and identi fication of functional genes related to sexual differentiation, gonad maturation and gametogenesis. For these reasons,we performed transcriptomic analysis on spotted scat using a multiple tissue sample mixing strategy. The Illumina RNA sequencing generated 118 510 486 raw reads. After trimming, de novo assembly was performed and yielded 99 888 unigenes with an average length of 905.75 bp. A total of 45 015 unigenes were successfully annotated to the Nr, Swiss-Prot, KOG and KEGG databases. Additionally, 23 783 and 27 183 annotated unigenes were assigned to 56 Gene Ontology (GO) functional groups and 228 KEGG pathways,respectively. Subsequently, 2 474 transcripts associated with reproduction were selected using GO term and KEGG pathway assignments, and a number of reproduction-related genes involved in sex differentiation,gonad development and gametogenesis were identi fied. Furthermore, 22 279 simple sequence repeat (SSR)loci were discovered and characterized. The comprehensive transcript dataset described here greatly increases the genetic information available for spotted scat and contributes valuable sequence resources for functional gene mining and analysis. Candidate transcripts involved in reproduction would make good starting points for future studies on reproductive mechanisms, and the putative sex differentiation-related genes will be helpful for sex-determining gene identi fication and sex-speci fic marker isolation. Lastly, the SSRs can serve as marker resources for future research into genetics, marker-assisted selection (MAS) and conservation biology.
Keyword: Scatophagus argus; illumina RNA-seq; reproduction; simple sequence repeat (SSR)
1 INTRODUCTION
Spotted scat ( Scatophagus argus), which has the notable ability to tolerate a wide range of environmental salinities, is widely distributed around the Indo-Paci fic region, including Southeast China (Barry and Fast,1992; Gupta, 2016). Spotted scat with such features as high nutritional value, easy cultivation, low feeding cost, etc. is increasingly becoming an important farmed fish with considerable economic value in East and Southeast Asia, and the commercial demand for seedlings is rising sharply. However, it is difficult to obtain spotted scat offspring via arti ficial breeding because of the different gonad maturation time between the sexes due to protandry (Chen et al.,2015a). Arti ficial reproduction remains a barrier for the aquaculture industry at present and juveniles still have to be captured from the wild. In the last few years, the natural population of spotted scat has been clearly declining (Gupta, 2016), so the quantity of available wild seedlings is severely limited.
A perfect arti ficial reproduction technology is the primary prerequisite for large-scale commercial fish breeding and culture (Xie et al., 2014). So far, a few studies have been carried out on induced breeding(Barry et al., 1991, 1993; Cai et al., 2010), reproductive biology (Cai et al., 2010; Zhang et al., 2013; Gandhi et al., 2014) and regulatory genes (Deng et al., 2014;Chen et al., 2015a, b; Li et al., 2015; Liu et al., 2015;Chen et al., 2016) in spotted scat, but most molecular biology studies have been based on a single-gene strategy. The mechanisms of reproductive development and regulation have been little investigated and are poorly understood. Therefore, it is necessary to elucidate the molecular mechanism of the reproductive process and identify more functional genes related to sex differentiation, gonad maturation and gametogenesis. Unfortunately, compared with other fishes with economic value, the genetic and genomic information available for spotted scat is rather limited.To date, just 304 DNA and RNA sequences and one SRA have been deposited in GenBank, including 24 ESTs and 13 mitochondrial genes (analyzed on February 15th, 2017). For these reasons, there is an urgent need for more genomic background knowledge and breeding programs to maintain the sustainable development of spotted scat fishery.
With the aid of next-generation high-throughput sequencing technologies such as the Illumina/Solexa platform, transcriptome sequencing has been in widespread use to obtain large-scale transcript sequences and gene expression data rapidly and costeffectively for non-model species without reference genomic information (Garber et al., 2011; Finseth and Harrison, 2014). Transcriptome analysis has been widely employed in genetics, molecular biology,ecology, and physiology, and plays signi ficant roles in gene discovery, gene expression and regulation analysis, and molecular marker development (Grabherr et al., 2011). Previous studies on Scophthalmus maximus (Ma et al., 2016), Trachinotus ovatus (Xie et al., 2014) Poec i lia reticulata (Fraser et al., 2011) and Macrobrachium nipponense (Ma et al., 2012) suggest that, transcriptome analysis is an extremely efficient approach to identify large numbers of functional genes and simple sequence repeat (SSR) markers. By transcriptome sequencing with the 454 pyrosequencing technology, Li et al. (2014) identi fied and validated 33 novel single nucleotide polymorphisms (SNPs) for population and conservation genetics of spotted scat,but did not conduct functional gene discovery and expression analysis.
Using the Illumina paired-end sequencing technology, high-through-put RNA sequencing, de novo assembly, functional annotation and classi fication were performed in the present study.This work is the first comprehensive transcriptome analysis on spotted scat based on a multiple tissue sample mixing strategy. This study was designed i) to enrich the available genetic and genomic information for a deeper understanding of gene expression,functional gene mining and SSR marker development,and ii) to identify putative genes related to reproductive processes such as the GnRH-GtH axis, sex differentiation, gonad maturation and gametogenesis for future research into the molecular mechanism of reproduction. These data are also an important resource for reproduction-related studies in other teleost fishes.
2 MATERIAL AND METHOD
2.1 Animal materials and sample preparation
One-year-old adult spotted scat (females: n=10;males: n=10) were collected from the Zhuhai Yucheng Fry Cultivation Base (Zhuhai, Guangdong, China),and sacri ficed by decapitation following MS222 anesthetization. Tissue samples including brain,pituitary, heart, muscle, head kidney, kidney, liver,gut, spleen, gill, testis and ovary were excised as soon as possible, frozen in liquid nitrogen immediately,and stored at -80°C until RNA extraction.
2.2 RNA extraction
Total RNA was extracted from each tissue of the females and males using Trizol Reagent (Life Technologies, Carlsbad, CA, USA) following the manufacturer’s instructions. The concentration of total RNA was determined by the absorbance at 260 nm using UV-spectrophotometry (Nanodrop,2000c, Thermo Scienti fic, Wilmington, DE, USA)and the quality was tested using OD260/280(1.8–2.0).Then, ethidium bromide staining of the 28 S and 18 S ribosomal bands on a 0.8% agarose gel was used to assess the RNA integrity.
2.3 Construction of cDNA libraries and sequencing
Two total RNA mixtures (approximately 5 μg)from the male and female tissues respectively were delivered to Gene Denovo Biotechnology Co. Ltd.(Guangzhou, China) for the construction of male and female cDNA libraries. In brief, the mRNAs were extracted from total RNA using an Oligo-dT Beads Kit (Qiagen, Hilden, Germany) and fragmented with fragmentation buffer. Then, double-stranded cDNA was synthesized using 200–700 nt fragments as templates, random primers, and a SuperScript doublestranded cDNA synthesis kit (Invitrogen, California,USA). Ligated fragments were then generated by a series of reaction processes including puri fication of PCR products, end repair, dA-tailing, and ligation of Illumina adapters. After agarose gel electrophoresis,suitable fragments were isolated and puri fied for PCR ampli fication and the cDNA libraries of the two sex groups were sequenced on an Illumina HiSeq™ 2000 platform (Illumina, Inc., San Diego, USA).
2.4 Trimming and de novo assembly
Quality control was performed by the SOAPnuke v1.5.0 software with the filter parameters “-l 10 -q 0.5-n 0.05 -p 1 -i”. The raw reads were filtered to generate high quality data via processes including removal of adapter sequences, reads with ambiguous sequences(N) more than 10% and low-quality sequences with percentage of low quality bases of quality value_5 was >50% in one read (base quality score <Q20). De novo assembly of the clean reads was carried out using Trinity software (version: r20140717, http://trinityrnaseq.sourceforge.net/) with default parameters. Trinity combines three components:Inchworm, Chrysalis and Butter fly. Firstly, Inchworm assembles reads using a greedy k-mer based approach,resulting in a collection of linear contigs. Next, in Chrysalis, the abundant contigs produced by the repeated Inchworm processes are built into de Bruijn graphs through k-1 overlaps. In the third phase,Butter fly, the fragmented de Bruijn graphs are trimmed, compacted and reconciled to final linear transcripts (Grabherr et al., 2011). Finally, redundant sequences are eliminated, and the longest transcripts are de fined as unigenes.
2.5 Functional annotation and classi fication
Functional annotation was performed by sequence comparison against public databases using BLASTx v2.2.23 (Camacho et al., 2008). All unigenes were searched against the Nr, Swiss-Prot, KOG and KEGG databases with an E-value cut-off of 1E-5. Further analysis of the annotation results was conducted using the Blast2GO software (Conesa et al., 2005) to obtain GO functional information. The WEGO statistical software was used to assay the GO functional results for GO term classi fication and visualization (Ye et al.,2006). In addition, the KEGG annotations were analyzed by using KOBAS v2.0 software for KEGG pathway categories (Kanehisa et al., 2004; Xie et al.,2011).
Meanwhile the King showed the Princess inside all his gold wares, every single bit of it--dishes, goblets, bowls, the birds and game, and all the wonderful beasts
2.6 SSR loci detection
The MIcroSAtellite software package (MISA) was employed to identify SSR loci in the assembled unigene sequences (Thiel et al., 2003). The searches were carried out with a minimum of six contiguous repeat units for di-nucleotide motifs and five for all other motifs. Mono-nucleotide repeats (MNRs) were excluded because of difficulties in distinguishing genuine MNRs and a few MNRs generated by sequencing errors. Primer pairs flanking each SSR locus were designed using the Primer 5.0 software(Lalitha, 2000).
3 RESULT
3.1 Illumina sequencing and assembly
RNA-seq produced 118 510 486 raw reads with an average read size of 125 bp, encompassing 14.81 Gb sequencing data (Table 1). Quality control of the raw data resulted in 116 588 098 clean reads, which is equal to 14.57 Gb sequencing data. The Q20 and GC percentage were 98.65% and 48.45%, respectively.The high-quality clean reads were de novo assembled subsequently and generated 99 888 unigenes with a mean length of 905.75 bp and an N50 length of 1 879 bp.The unigene lengths ranged from 201 to 16 393 bp. In detail, 42 439 (42.49%) unigenes were >500 bp in length, 23 868 (23.89%) were >1 000 bp and 12 289(12.30%) unigenes were >2 000 bp in length (Fig.1).
3.2 Functional annotation and classi fication
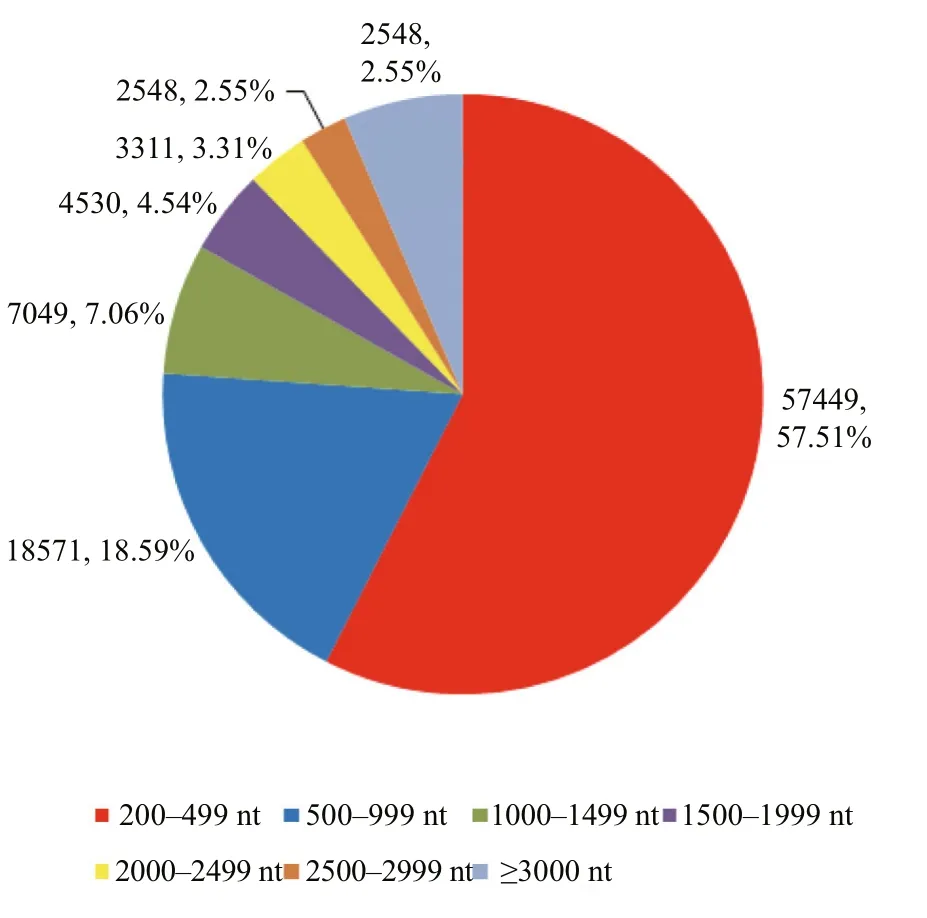
Fig.1 Sequence size distribution of all assembled unigenes derived from the spotted scat transcriptome

Table 1 Statistics for the sequencing and assembly of the spotted scat transcriptome
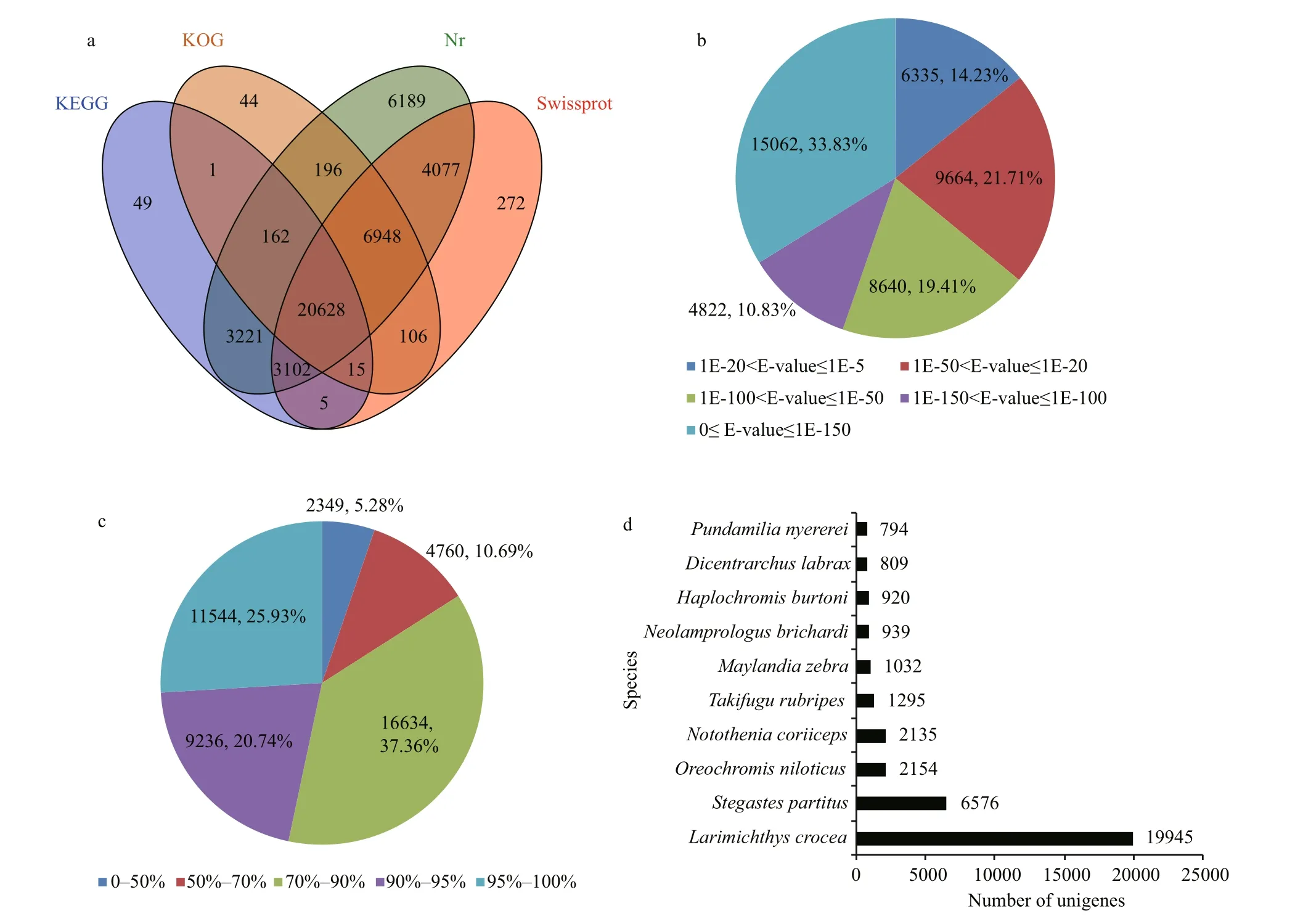
Fig.2 Statistical summary of unigene annotation
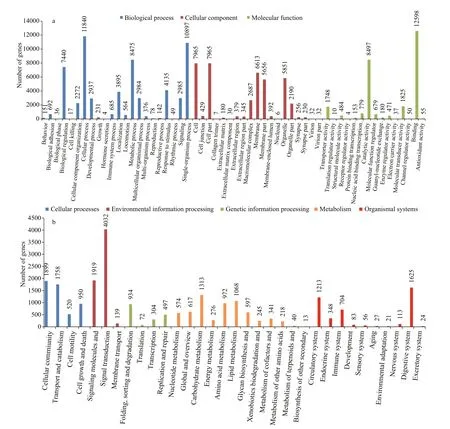
Fig.3 Gene Ontology (GO) and KEGG pathways functional classi fication of annotated unigenes
Among all the assembled unigenes, 44 523(31.41%), 35 153 (27.00%), 28 100 (28.13%) and 27 183 (27.21%) unigenes showed signi ficant similarity to proteins in the Nr, Swiss-Prot, KOG and KEGG databases, respectively (Fig.2a). A total of 20 628 unigenes were annotated in all four databases,but 54 873 (54.93%) unigenes could not be annotated in any of them. The E-value distribution showed that 64.07% of unigenes exhibited signi ficant homology(below 1E-50) in the Nr database and 21.71% of homologous sequences ranged from 1E-20 to 1E-50(Fig.2b). Moreover, 84.03% of the annotated unigenes displayed more than 70.00% similarity (Fig.2c).These results con firmed that the transcriptome data were successfully annotated. The homologous genes came from 370 species, and the top five species showing BLASTx hits were Larimichthys crocea(19 945, 44.80%), Stegastes partitus (6 576, 14.77%),Oreochromis niloticus (2 154, 4.84%), Notothenia coriiceps (2 135, 4.80%) and Takifugu rubripes(1 295, 2.91%) (Fig.2d). This result suggested that spotted scat is closely related to L. crocea, whose genome has been completely sequenced and published(Xiao et al., 2015).
After Blast2GO analysis, a total of 23 783 annotated unigenes in the GO database were assigned to 56 functional groups (Fig.3a). Of which, 60 885 (46.94%)transcripts comprised the largest category of‘biological process’, followed by ‘cellular component’(41 245; 31.80%) and ‘molecular function” (27 570;21.26%). The GO terms ‘cellular process’ (11 840;19.45%), ‘cell’ (7 965; 19.31%) and ‘binding’ (12 598;45.69%) were the largest groups in the three respective main categories. According to KEGG pathway analysis, 27 183 (27.21%) unigenes were assigned to five main categories, which consisted of 228 different pathways (Fig.3b). Among the five main categories,‘metabolism’ pathways were the most abundant group with 6 274 (26.15%) genes, followed by‘environmental information processing’ (6 090;25.38%), ‘cellular processes’ (5 127; 21.37%),‘organismal systems’ (3 866; 16.11%) and ‘genetic information processing’ (2 639; 11.00%).
3.3 Identi fication of reproduction related genes
GO terms and KEGG pathways associated with reproduction were selected to identify candidate sequences with important reproductive functions. As a result, 604 and 1 870 transcripts annotated with reproduction-related GO terms and KEGG pathways were obtained, respectively (Table 2). GO terms under ‘reproduction’, ‘reproductive process’,‘hormone activity’, ‘receptor binding’ and ‘receptor activity’ were selected. The reproduction-related pathways found in KEGG analysis were ‘GnRH signaling pathway’, ‘progesterone mediated oocyte maturation’, ‘estrogen signaling pathway’, ‘oocyte meiosis’, ‘steroid biosynthesis’, ‘steroid hormone biosynthesis’, ‘retinol metabolism’, ‘neuroactive ligand receptor interaction’ and ‘neurotrophin signaling’. Details of these candidate reproduction related genes are given in Supplementary Table 1.
Then, transcripts showing homology to genes associated with the gonadotropin-releasing hormone(GnRH)-gonadotropic hormone (GtH) axis, gonadal differentiation and maturation, steroid hormone biosynthesis, gametogenesis and gametic function were identi fied (Table 3). The most represented gene products included GnRH receptor (GNRHR), GtH,follicle-stimulating hormone (FSH), Lutropinchoriogonadotropic hormone receptor (LHCGR),doublesex and mab-3 related transcription factor(DMRT), anti-Müellerian hormone (AMH), protein fem-1 homolog (FEM1), forkhead box l2 (FOXL2),wingless-type MMTV integration site family member-4 (WNT4), cytochrome P450 family members (CYPs), StAR-related lipid transfer protein(STARD), vitellogenin (VTG) and spermatogenesis-associated protein (SPATA). These gene sequences could provide valuable candidates for further functional analysis and future studies on the reproduction mechanism and its regulatory networks.

Table 2 Candidate transcripts related to reproduction identi fied from GO terms and KEGG pathways
3.4 Discovery and characterization of SSR loci
Using the MISA program, a total of 15 700(15.72%) unigenes containing 22 279 SSRs were preliminarily identi fied, with a distribution density of one SSR per 4.06 kb (Table 4). Among these unigenes,4 281 contained more than one SSR. The top five most abundant motifs were di-nucleotide (13 229;59.38%), tri-nucleotide (6 022; 27.03%), tetranucleotide (2 330; 10.46%), penta-nucleotide (509;2.28%) and hexa-nucleotide repeats (189; 0.85%). A frequency distribution analysis of the SSRs based onthe motif sequence types was conducted (Fig.4). The spotted scat transcriptome was rich in AC/GT (9 563;42.92%), AG/CT (2 548; 11.44%) and AGG/CCT(1 852; 8.31%) repeats. The SSR primer pairs are given in Supplementary Table 2.
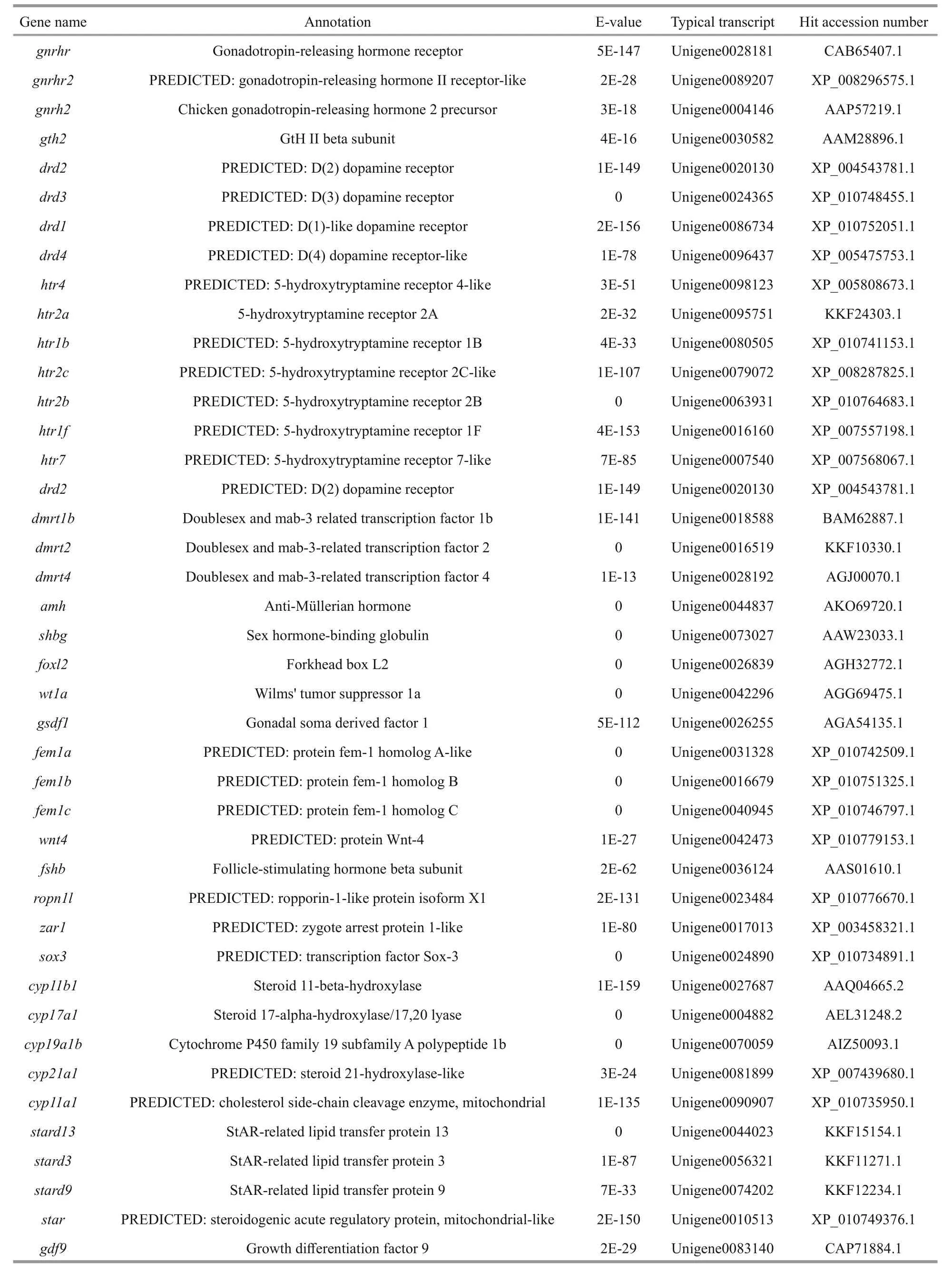
Table 3 Reproduction related genes found in the spotted scat transcriptome

Table 3 Continued
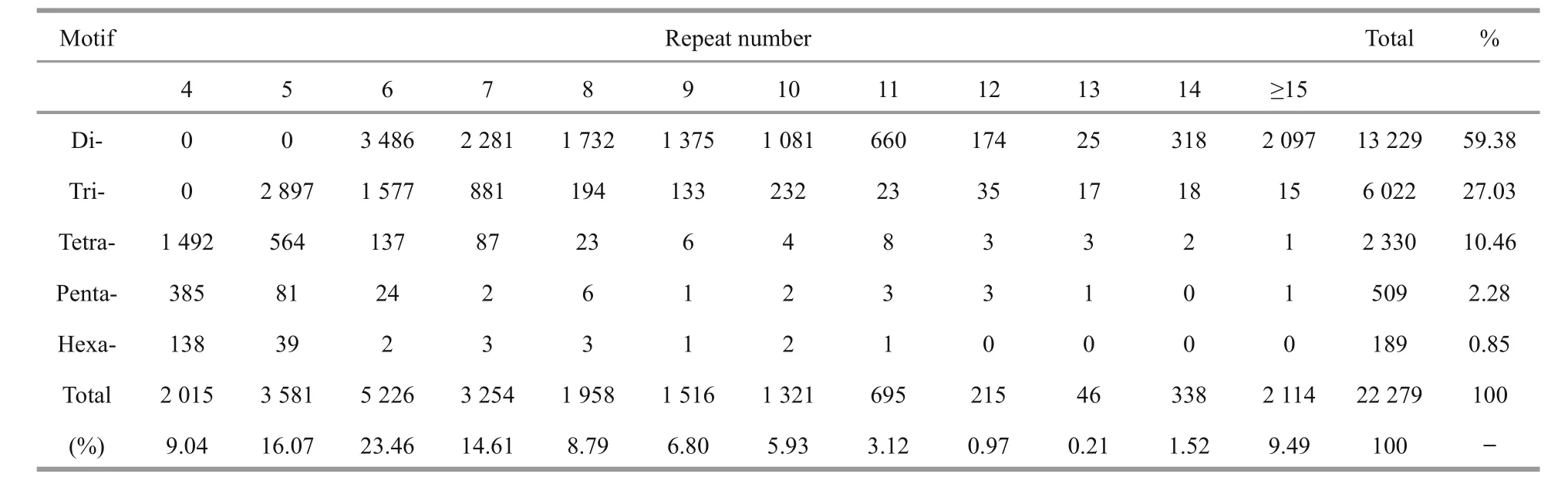
Table 4 Summary of the motif distribution of SSR loci identi fied in the spotted scat transcriptome

Fig.4 Frequency distribution of classi fied repeat types of SSR loci identi fied in the spotted scat transcriptome
4 DISCUSSION
4.1 Sequencing, assembly and annotation
The BLASTx top-hit species distribution showed that approximate half of the homologous genes came from L. crocea (Fig.2c). As one of the most economically important farmed fish species, the whole genome of L. crocea has been completely sequenced and published (Xiao et al., 2015).Therefore, it is not surprising that S. argus had a signi ficantly higher number of similar sequences to L. crocea, in comparison with the other matched species without genomic information. Notably, most of the unigenes were matched to L. crocea, rather than other teleost fishes with whole-genome sequence data such as Cynoglossus sem i laevis (Pleuronectiformes),Danio rerio (Cypriniformes), and Ctenopharyngodon idellus (Cypriniformes). These results suggest the gene content is phylogenetically conserved between S. argus (Perciformes: Scatophagidae) and L. crocea(Perciformes: Sciaenidae), and that S. argus is more closely related to L. crocea, which also belongs to the order Perciformes, than to the other sequenced teleost fishes.
4.2 Reproduction-related genes
Fish reproductive activities are basically manipulated by the GnRH-GtH axis, as GnRH acts on pituitary gonadotropes to regulate the synthesis and release of LH and FSH (Levavi-Sivan et al., 2010;Prasad et al., 2015). We identi fied some transcripts homologous to GNRHR, GtH2, FSH, FSH receptor and LHCGR, which are considered to be critical regulators or factors in sex hormone secretion and gonad maturation, in the transcriptome data (Table 3).GnRH agonists and exogenous GtH preparations have been widely used to induce ovulation and spermiation in farmed fish (Xie et al., 2014). To date,LHRHa (Luteinizing Hormone Releasing Hormoneanalog), LHRH-A2, 17α, 20β-dihydroxyprogesterone,Human Chorionic Gonadotropin (HCG), DOM(Domperidone) and methyl testosterone have been used in attempts to induce breeding in spotted scat(Gupta, 2016). Nevertheless, a practical arti ficial breeding technology has not been established. A lack of understanding of regulatory mechanisms leads to the abuse of hormones in induced spawning practices.Hence, intensive investigation on the GnRH-GtH axis is the principal task for successful arti ficial breeding.In addition, several former studies have shown the involvement of serotonin (5-HT) and dopamine in the regulation of the GnRH-GtH axis (Levavi-Sivan et al., 2012; Prasad et al., 2015). It is very interesting that we also found diverse 5-HT and dopamine receptors in spotted scat (Table 3). All of these GnRHGtH axis-speci fic sequences will greatly help to elucidate the regulatory mechanisms of the reproductive axis and improve preparation development in future research.
The major objective of induced breeding is to promote gonadal development and produce mature oocytes and motile sperms. We recognized several unigenes associated with oogenesis and oocytes maturation, such as VTG and zona pellucida spermbinding protein. VTG has a greater transcript abundance in the ovary compared with the testis and is an important protein for oocyte maturation (Patnaik et al., 2016). Zhang et al. (2013) found that formulated diets with supplementation of fish oil could effectively increase spotted scat liver vtg expression and promote ovarian development. Furthermore, we identi fied transcripts homologous to SPATA, Vasa, sperm flagellar protein, spermine oxidase and spermidine synthase which are potentially associated with spermatogenesis and sperm motility. Overall, in order to increase hatchability and the survival ratio, further studies focused on reproductive success and establishing highly effective techniques for the induction of gamete maturation, ovulation, and spermiation are highly encouraged.
Androgen and estrogen play critical roles via androgen receptors (ARs) and estrogen receptors(ERs) in vertebrate reproductive systems. Several types of ER but only one type of AR were found in our data (Table 3). This result indicates that spotted scat has high diversity in estrogen receptor signaling.Chen et al. (2016) reported that ARs were expressed in both male and female spotted scat and showed obvious tissue-speci fic expression patterns. This finding implies that ARs are multifunctional in different tissues and that appropriate AR expression in the ovary is necessary for proper ovarian development.
The development of ovaries and testes from undifferentiated gonads is regulated by sex differentiation related genes (Ma et al., 2016).Transcripts homologous to genes involved in the control of sex differentiation were discovered, such as DMRT family members, FOXL2, WNT4, CYPs,AMH and FEM1 (Table 3). Cyp 11 a 1, cyp 11 b 1,cyp 17 a 1, cyp 19 a and cyp 21 a 1 have crucial functions in the biosynthesis of sex steroid hormones, which are generally regarded as central to sex regulation.Moreover, foxl 2 is one of the most conserved genes involved in the differentiation and maintenance of ovaries in vertebrates, and generally participates in ovarian development as the activating transcription factor of cyp 19 a (Ma et al., 2016). In ovaries and testes of spotted scat, foxl 2 and cyp 19 a 1 exhibited coordinate expression patterns at different development stages (Liu et al., 2015), suggesting that foxl 2 may regulate cyp 19 a 1 expression.Coincidentally, there was a signi ficant positive correlation between foxl 2 mRNA and serum 17β-estradiol levels (Li et al., 2015). WNT4 plays a crucial role in ovarian differentiation and development in mammals but its function in teleost fish remains unclear. Two wnt 4 subtypes, wnt 4 a and wnt 4 b, have been identi fied in spotted scat. W nt 4 a might be involved in gonad development and play a role in spermatogonial proliferation, while wnt 4 b might only be involved in ovary development (Chen et al., 2016).Sex-determining region Y box 9 ( sox 9) is an important male sex-determination gene associated with testis development in vertebrates (Morrish and Sinclair,2002). We found sox 3 in our data but sox 9 was absent,possibly because sox 9 expression is signi ficantly downregulated after the completion of gonadal differentiation. In accordance with this prediction,sox 9 expression was signi ficantly up-regulated in the early stages of masculinization induced by an aromatase inhibitor (Chen et al., 2015b), indicating that sox 9 might be one of the most pivotal factors involved in sex determination in spotted scat.
Because of their markedly faster growth rate,female monosexual farming would be more efficient in spotted scat fishery (Chen et al., 2016). Because spotted scat fry have no obvious sexual morphological differences, sex-speci fic molecular markers are a prerequisite for sex identi fication in fry breeding. As thus, studying the roles of sex differentiation-related genes in sex determination will greatly help to isolate sex-speci fic markers (Xie et al., 2014). DMRT1 was con firmed to be the master regulator of male gonad differentiation and has attracted considerable interest(Herpin and Schartl, 2011). Three DMRT family members, dmrt 1 b, dmrt 2 and dmrt 4, have been recognized, but their sex-regulating effects in spotted scat are still unknown. Additionally, the functions of sex-related genes such as amh, fem 1 and wt- 1 have also been rarely investigated and are poorly understood. Hence, further molecular biology and biochemical studies are required to clarify the sexual manipulation roles of these genes and con firm the sex determining gene.
4.3 SSR loci discovery
SSRs, tandemly repeated motifs with high-levels of polymorphism, are widely used as molecular markers in population genetics, conservation genetics,genetic linkage mapping and quantitative trait locus analysis (Patnaik et al., 2016). SSR repeat types generally exhibit signi ficant species-speci fic differences in fish, molluscs and crustaceans (Jung et al., 2011; Ma et al., 2012; Tong et al., 2015). The most abundant loci in spotted scat were di-nucleotide (AC/GT, AG/CT) and tri-nucleotide repeats (AGG/CCT)(Fig.4). The Cristaria plicata transcriptome is also rich in AC/GT (28.94%) motifs (Patnaik et al., 2016),but has a much lower ratio than that in spotted scat.Furthermore, the most frequent motifs among both diand trinucleotide repeats were obviously different from those in T. ovatus, in which AT/GT and ACG/CTG accounted for 43.5% and 10.2% of motifs,respectively (Xie et al., 2014). SSR markers have become an effective approach to improve the production of commercially important fish species(Liu et al., 2013). To date, a few polymorphic SSR makers have been developed in spotted scat (Liu et al., 2010, 2013), but the amount is still far from adequate. The SSR loci identi fied here could be a powerful tool for future studies on genetics and MAS as potential molecular markers.
5 CONCLUSION
In this study, we used Illumina high-throughput sequencing to analyze the transcriptome of spotted scat. The transcript dataset presented here greatly increases the genetic information available for general gene expression studies and contributes valuable sequence resources for functional gene excavation and analysis. A number of candidate functional genes involved in reproduction processes would make good starting points for future studies on the regulatory mechanisms of the GnRH-GtH axis, sex differentiation, gonadal development, gametogenesis,oocyte maturation and sperm motility. The sex differentiation-related transcripts we identi fied will be useful for identifying sex-determining genes and isolating sex-speci fic markers. Finally, the massive amount of SSRs we generated will provide abundant marker resources for future research into genetics,MAS and conservation biology.
6 AUTHOR CONTRIBUTION
Conceived and designed the experiments: HC and WY. Performed the experiments: XC and KZ.Analyzed the data: WY and HC. Contributed reagents/materials/analysis tools: DJ, SD and CZ. Wrote the manuscript: WY and HC. Overall monitoring,manuscript overview and editing: GL and CZ.
7 DATA AVAILABILITY STATEMENT
The original Illumina HiSeq sequencing data are available from the NCBI Sequence Read Archive(SRA) database under the accessions SRX2499155(male) and SRX2499156 (female). Other data that support the findings of this study are available from the corresponding author upon reasonable request.
猜你喜欢
杂志排行

Journal of Oceanology and Limnology的其它文章
- Editorial Statement
- Recent insights into physiological responses to nutrients by the cylindrospermopsin producing cyanobacterium,Cylindrospermopsis raciborskii*
- Response of Microcystis aeruginosa FACHB-905 to different nutrient ratios and changes in phosphorus chemistry*
- In fluence of light availability on the speci fic density, size and sinking loss of Anabaena flos- aquae and Scenedesmus obliquus*
- Application of first order rate kinetics to explain changes in bloom toxicity—the importance of understanding cell toxin quotas*
- Regime shift in Lake Dianchi (China) during the last 50 years*