土壤样品中重金属化学形态模型的发展与应用
2018-08-02邓迎璇李永涛李晓晶陈雅丽翁莉萍
邓迎璇,李永涛,2,李晓晶,王 龙,马 杰,陈雅丽,翁莉萍*
(1.农业部农产品质量安全环境因子控制重点实验室/农业部环境保护科研监测所,天津 300191;2.华南农业大学资源环境学院,广州 510642)
土壤样品中的(类)重金属元素[如镉(Cd)、砷(As)]以多种化学形态存在,重金属在土壤中的生物有效性、生物毒性和迁移性,不但受其总浓度的影响,也取决于其化学形态分布。因此了解和预测土壤样品中重金属的化学形态分布,对提高其环境风险评估与管控的准确性与有效性,从而保持生态系统的可持续发展十分重要。
大部分土壤中的重金属存在于固相中,主要包括:存留于(黏土)矿物颗粒中的杂质、由沉淀作用等形成的难溶解态物质、吸附在各种颗粒表面的吸附态重金属;液相介质中的重金属形态主要有:自由态离子、与无机配体结合态、与溶液中的(有机和无机矿物)胶体颗粒物结合的吸附态重金属。溶液中重金属的各种形态与其元素在环境中的迁移性密切相关。一般认为,溶液中的自由态离子是能被植物根系直接吸收的化学形态,由此产生了“自由离子活度模型”(Free Ion Activity Model)[1-2]。存在于固相中的吸附态重金属和部分易于溶解的沉淀态重金属,由于能较为快速地释放到溶液中,而被称为具有反应活性(Reactive)的化学形态。相反,(黏土)矿物结构内部和难以溶解的沉淀中的重金属,由于短期内不能释放,而被称为惰性或没有反应活性(Non-reactive)的化学形态。由此可见,重金属的化学形态受吸附、络合和沉淀等反应过程控制,而吸附反应在控制土壤样品中重金属的化学形态分布上起着重要作用。
研究土壤中重金属化学形态的方法主要有实验分析和模型计算两种。土壤样品中重金属的全量消解常采用含有氢氟酸的酸溶法和碱熔法,非全量或近全量消解主要采用强酸组合或者强酸与氧化剂的组合。除了土壤全量,重金属化学形态分析传统上主要采用具有不同强度和提取机制的提取剂进行振荡提取[3]。这些提取方法进行土壤重金属化学形态分析的优势在于其比较简便易行,但是存在专一性差、形态解释与实际不符、提取过程中发生再分配等问题[4]。为了能更好地评估土壤中重金属的生物有效性,人们又发明了诸如同位素稀释[5]和梯度扩散薄膜技术(Diffusive Gradients in Thin-films,DGT)等含有动力学意义的形态测定方法[6-7]。仅少数分析方法可以直接测定溶液中重金属离子形态,如离子选择电极、(离子交换-)高压液相色谱、多南膜(Donnan Membrane Technique,DMT)技术等[8-11]。
应用化学分析方法测定元素化学形态具有直接、准确的优势,但是化学分析方法较为昂贵耗时,且现有的分析方法不能给出一种重金属在土壤样品中分布的全貌,常用的分析方法并不具备识别化学形态的特异性。比如:环境中普遍存在的天然有机质(Natural Organic Matter,NOM)能够结合金属阳离子如Cd2+,形成NOM络合态Cd,但是测定这些结合态的金属含量在方法上难以实施。而且,化学分析方法测得的元素化学形态仅适用于当前测定的样品,一旦更换研究体系,分析结果很可能将不再适用,因此化学测定分析方法的预测性通常较差。为了克服以上化学分析方法中的缺点,人们开发了利用模型计算与预测元素化学形态的手段。本文综述了用于土壤环境样品中重金属元素化学形态模型的发展,介绍了几种机理性吸附模型的主要机制,总结了用于预测重金属离子在天然环境样品上吸附模型参数的获取方法,具体模型应用的方法以Cd和As分别作为环境中金属阳离子和含氧阴离子的代表进行了阐述,并对模型的应用和发展进行了展望,以期为今后化学模型用于元素在天然环境样品中化学形态分布的预测提供借鉴。
1 经验性模型与机理性模型
1.1 经验性模型
环境样品中重金属的化学形态模型可分为经验性模型和机理性模型。经验性模型是基于实际样品的实验室分析数据,通过回归分析等统计手段建立起来的某种化学形态的浓度或元素分配系数与其他因子的数量关系。对于土壤样品,此类模型也被称为土壤传递函数(Pedo-transfer function)[12]。
最简单的经验性模型是线性(吸附)固-液分配模型:Q=KdC(其中:Q为某种重金属在固相中的含量,mg·kg-1);Kd为固-液分配系数,L·kg-1;C为该元素在溶液中的浓度,mg·L-1)。一般情况下,线性关系只有在重金属浓度很低的情况下才成立,而且在不同土壤中Kd值也会随着重金属含量、pH、土壤有机质(SOM)含量等因素而变化,其数值变化可能跨越几个数量级[13-15]。但是为了便于计算,在很多涉及元素固-液分配的生物地球化学模型中,人们仍经常采用单一固定的Kd值,这给模型预测的准确性带来很大影响。比线性模型略微复杂、适用性更强一些的是Freundlich 模型[16-17]:Q=KFrCn(其中:KFr是吸附/反应常数,其单位取决于参数n的数值;n没有单位,其数值一般<1,当n=1时Freundlich模型等同于线性模型)。与线性模型相比,Freundlich模型考虑重金属固-液分配的非线性表现,其适应的重金属浓度范围更加广泛。除了浓度之外,土壤pH等因素也影响到重金属元素在土壤中的化学形态分布[18]。为了提高模型的普适性,增加了其他因子的扩展,Freundlich模型被广泛应用。将Freundlich模型进行对数转换可将模型转换为线性关系。
类似于Freundlich模型的对数线性经验性模型被广泛应用于土壤中重金属离子(如:Cd2+、Cu2+、Pb2+)化学形态的计算[13,19-20],但针对同种元素不同研究得到的模型考虑的因子有所不同。以Cd为例,在Luo等[19]、Sauve等[13]和 Mcbride等[20]的研究中用于表述土壤溶液中Cd浓度的经验性模型分别采用了1、2、3个因子作为自变量,公式(1)~公式(3):
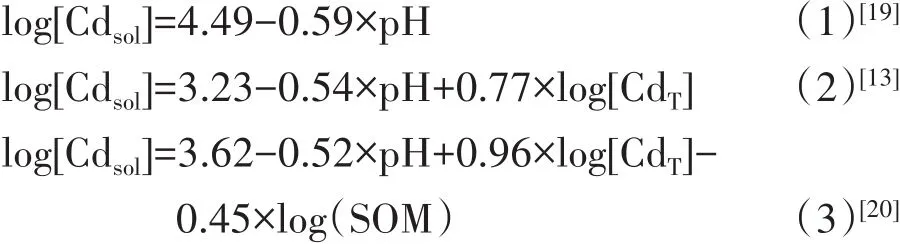
式中:Cdsol为Cd在土壤溶液中的浓度,μg·L-1;CdT为土壤中Cd的总量,mg·kg-1;SOM为土壤有机质含量,%。之所以选择了不同数量的自变量是受不同作者对模型计算的简便性和准确性之间权衡的影响,也受到所用数据中不同因子的变化范围和土壤性状等其他因素的影响。Luo等[19]研究显示溶液中的Cd与土壤体系pH呈线性相关[公式(1)],加入SOM并不能显著提升模型拟合的准确性。另外,即使模型拟合选用相同的自变量,不同文献中模型参数数值之间通常也存在较大差异。同样以Cd为例,Sauve等[13]和Hao等[21]都采用了pH和土壤总Cd含量来计算溶液中的Cd浓度,如公式(2)和(4)所示:
log[Cdsol]=-1.515+0.072×pH+0.685×log[CdT](4)[21]
可以看出,同样是采用pH和CdT这两个变量因子,但所对应的模型参数相差很大,甚至代表因子影响方向的参数正负符号都可能相反[13,21]。由于模型的经验性,数据范围和主要限制因子的变化可能导致拟合的模型参数的不同。
由以上实例可见,经验性模型具有计算便捷和易于理解掌握等优点,所取得的模型及其参数在一定范围内能够很好地表述重金属离子在固-液介质中的分配,同时也能在一定程度上揭示可能的控制机制。但是,经验性模型是“黑箱模型”,受模型的统计经验性制约,模型的使用局限于建模时所采用的参照数据范围,而土壤体系组分和溶液化学性质的多变性妨碍了这些经验性模型在不同环境样品中的应用,致使经验性模型具有普适性差、机理性不清等缺点。因此经验模型方法在计算重金属离子形态上的不足逐渐被人们意识到[13-15,22]。
1.2 机理性模型
土壤中重金属化学形态的机理性模型是基于对反应过程的机制分析,应用化学反应定律而建立的模型,亦即“白箱模型”。瑞士水化学家Werner Stumm在研究中引入了化学平衡的概念,推动了基础化学原理在定量计算环境样品中化学反应过程的应用[23],Stumm相信要深入理解地方、区域和全球性的元素循环和污染影响,分子水平的信息是必不可少的。这一理念也逐渐被土壤化学领域所接受与应用。有关酸-碱反应、配位化学、沉淀-溶解平衡,相转换过程、氧化-还原过程,表面化学、光化学原理、固体溶解速率和颗粒传输等过程的物理化学基本理论与计算方法被逐渐融入到水、土壤等环境介质定量化学模型中。
土壤中重金属化学形态机理性模型的建立也是基于化学平衡这一概念,基于热力学原理而构建的。根据土壤的基本组成特点,至少需要考虑:(1)溶液中的化学反应;(2)固-液间的沉淀-溶解过程;(3)溶质在固体表面的吸附-解吸过程。土壤溶液化学反应中大部分过程的计算方法比较成熟,如酸-碱反应采用水解离常数、化合反应采用化合常数、氧化-还原反应根据氧化-还原电位和氧化-还原常数、CO2的溶入应用Henry常数等,所需参数也比较完备。固-液之间的沉淀-溶解反应的计算原理也比较简单,采用离子溶度积来计算,应用的难点在于沉淀-溶解过程是否达到最终的平衡。沉淀-溶解反应的进程较慢,即使溶质浓度达到超饱和状态,相应的沉淀也未必形成,或者先形成一种溶解度更高的沉淀、再逐步老化达到最终溶解度低的沉淀物质。土壤中含有多种有重金属吸附活性的颗粒,主要包括天然有机质、金属氧化物和黏土矿物。重金属离子通过化学键合(特异性反应)和静电作用(非特异性反应)吸附在这些颗粒表面,形成多种表面形态。离子的吸附不但受到颗粒表面特征的影响,也取决于pH、离子强度和其他吸附质的存在。如何准确计算离子在溶液中胶体表面和固相中颗粒表面的吸附过程是建立土壤重金属化学形态机理性模型的主要难点之一。
表面络合模型(Surface Complexation Model,SCM)是用于表征和计算离子等吸附质在吸附剂颗粒表面结合与富集过程的物理化学模型。常用的 Langmuir[24-25]模型[公式(5)]反映了表面位点的概念和位点饱和现象:

式中:KLa为吸附常数,L·kg-1;C为平衡体系中溶液中吸附质的浓度,mg·L-1;Qmax为最大吸附容量,mg·kg-1。尽管可以扩展为多吸附质竞争和多种位点模型,但只采用上述Langmuir模型还不能应对溶液化学组成变化对离子在带有永久/可变电荷吸附剂表面吸附的影响。将吸附反应过程分为特异性化学反应和非特异性静电作用是将表面络合模型机理化的重要一步,分别以位点与吸附质之间的络合反应模型和静电模型来表示。离子与颗粒表面的活性官能团可形成内圈配合物(离子直接与吸附剂表面官能团络合,形成的配合物之间没有水分子)和/或外圈配合物(离子与吸附剂表面官能团之间包含至少1个水分子)。表面络合反应的最基础数学表达式即为Langmuir模型[公式(5)],针对离子在铁、铝等金属(氢)氧化物颗粒表面吸附而发展的模型基本采用了Langmuir模型作为化学络合模型[26-32]。针对金属氧化物的表面络合模型的主要区别在于表面活性位点的类型和数量,以及采用的静电模型:扩散双电层模型(Diffuse Double Layer,DDL)、基本斯特恩模型(Basic Stern Model,BSL)和扩展斯特恩模型(Extended Stern Model,ESL)等。在这些模型中,CD-MUSIC(Charge Distribution and Multi-Site Complexation)模型[33-34]是较为先进的离子在氧化物上的表面络合模型。其位点种类和位点密度源于对矿物表面结构的分析,而离子在氧化物表面的化学形态尽量基于同步辐射技术[35-36]结构和分子动力学预测。光谱技术可以直接揭示离子吸附的微观机制,包括吸附离子在吸附剂表面形成的化学键数目,从而增加了模型的机理性和预测性。除微观的光谱分析外,宏观实验结果(如:电荷滴定曲线、离子强度对吸附的影响、反应过程热量的变化)亦能揭示离子吸附的机理。以离子强度对吸附的影响为例,离子吸附受体系离子强度变化的影响可以间接反映阳/阴离子在吸附界面形成配合物的类型[37-38]。
土壤有机质是土壤中吸附重金属离子的重要组分,其化学组成的复杂性和由此带来的表面位点的多样性为其表面络合模型的建立带来挑战。计算离子在有机质上的吸附时,常用到的两种先进的模型是:NICA-Donnan(Non-Ideal Competitive Adsorption)模型[39-40]和 WHAM(Windermere Humic-Aqueous Model)模型[41-42],这两种模型均考虑了有机质结合位点的异质性。其中WHAM模型[41-42]采用了离散的、非连续分布的处理,即将有机质上的吸附位点分为羧基类和酚羟基类两大类,每一类位点又分为四种,离子在这四种位点的吸附均以Langmuir模型表示,而同一类的四种位点的KLa值之间保持一定距离分布。NICA模型[39-40]将天然有机质上的羧基类和酚羟基类位点分别视为具有连续分布,其特异性吸附模型:
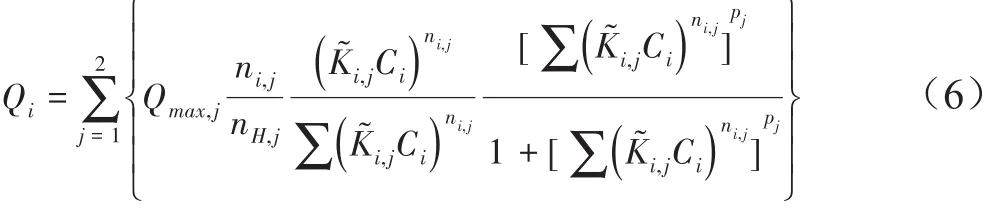
式中:Qi为有机质结合态i离子的含量,mol·kg-1;Qmax,j为有机质颗粒上 j类活性配体的总量,mol·kg-1;ni,j为 i离子结合的j类配体的特异性、非理想性参数(0< ni,j≤1);ni,j/nH,j描述的是离子i化学反应的平均计量系数;K͂i,j为平均亲和常数;Ci为离子 i在 Donnan相的浓度,mol·L-1;pj为 j类活性配体的化学异质性(0< pj≤1)[40]。NICA模型假设每一类位点的同一离子表面络合常数(K)是遵循接近正态分布(Sips分布)连续分布。WHAM模型和NICA模型,都结合了Donnan模型作为离子在天然有机质上吸附的静电模型。不同有机质的Donnan体积大小与腐植酸的性质及体系的离子强度等相关[43-44]。
表面络合模型(SCMs)在离子形态上预测的发展多经历从单一介质(金属氧化物、纯化腐植酸等)至天然环境样品(土壤、水体、沉积物)的过程。目前,针对单一介质的机理性模型的发展已经比较成熟。图1和图2分别为Cd在从土壤中纯化的胡敏酸(Humic Acid,HA)和富里酸(Fulvic acid,FA)上的吸附[40,45]及Cd、As在针铁矿上的吸附[46-47],并比较了实验数据与NICA-Donnan模型和CD-MUSIC模型的计算结果。可以看出,模型可以很好地描述实验数据。受益于模型的机理性特征,这些模型能够可靠计算在多种吸附离子存在情况下颗粒表面的相互协同或者竞争作用[48-49]。目前,针对代表天然有机质的HA和FA已经建立了较为完备的NICA-Donnan和WHAM的模型参数。针对常见的重金属离子在铁氧化物上的吸附,也有部分CD-MUSIC模型参数可以使用。
2 土壤中重金属化学形态机理模型的建立
2.1 建立土壤中重金属化学形态机理模型面临的挑战
基于物理化学的基本原理,从分子水平过程出发,建立土壤重金属化学形态的机理性模型,旨在提升模型的定量性、普适性和预测性。由于土壤体系的复杂性,模型的建立需综合考虑土壤溶液中的反应、沉淀-溶解和吸附反应等过程,而吸附反应部分的模型建立是土壤重金属化学形态模型发展的一个关键部分。如上所述,在过去研究中很多先进的表面络合模型的发展使得定量分析离子、小分子物质在胶体颗粒上的吸附得以实现[33,47,50-53]。但是,如何将模型预测由基于单一类型纯化的吸附剂的研究体系应用至环境样品分析面临诸多挑战:
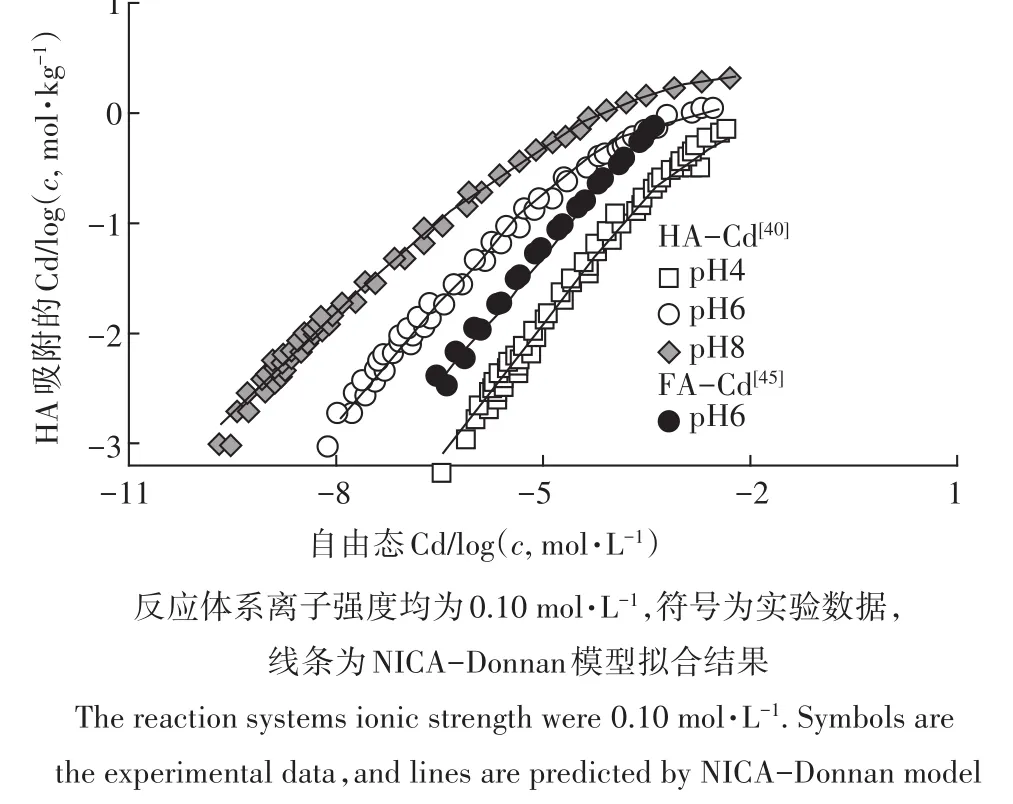
图1 镉在HA和FA上的吸附[40,45]Figure 1 Cd binding to HA and FA[40,45]
(1)需要选取某种胶体颗粒作为土壤中一类吸附界面的代表,并确定土壤中该类吸附界面的吸附能力相当于多少选定的模型胶体颗粒。如从土壤中提取的HA和FA常被选作代表土壤中具有重金属阳离子吸附能力的天然有机质模型胶体颗粒。但是不同土壤的有机质组成、反应活性存在差异[54],同时土壤有机质对重金属吸附能力也区别于用作模型材料的HA、FA;再如金属氧化物对重金属离子的吸附能力不仅取决于其在土壤中的含量,也取决于颗粒的比表面积,即矿物颗粒的粒径分布。(2)如何取得土壤中重金属的“活性总量”是建立土壤重金属化学形态机理模型面临的另一问题。因为土壤黏土矿物结构和难溶的其他矿物中的重金属在一般考虑的相关时间内不会参与反应,所以,在以化学平衡为前提的重金属化学形态模型计算中被视为“非活性部分”需要排除在外。而常用的强酸消解等方法得到的土壤重金属含量同时包含了“活性”和“非活性”部分的重金属。(3)面临的挑战还包括,离子在代表性胶体颗粒上吸附的研究大多采用单一类型颗粒,而土壤中同时含有多种类型的吸附介质,最直接的模型建立处理方式是假设各类别吸附介质之间没有交互作用,重金属在土壤中的吸附总量是在各类界面上吸附量的加和,这种线性叠加模式就是常用的多表面模型(Multi-Surface Model)的基本假设[55]。但是,天然环境样品中不同类型的颗粒之间可能存在着复杂的交互作用[56],如天然有机质与金属氧化物颗粒之间的交互作用会影响离子的吸附,导致离子总量不等同于在各类界面上的简单加和[55]。
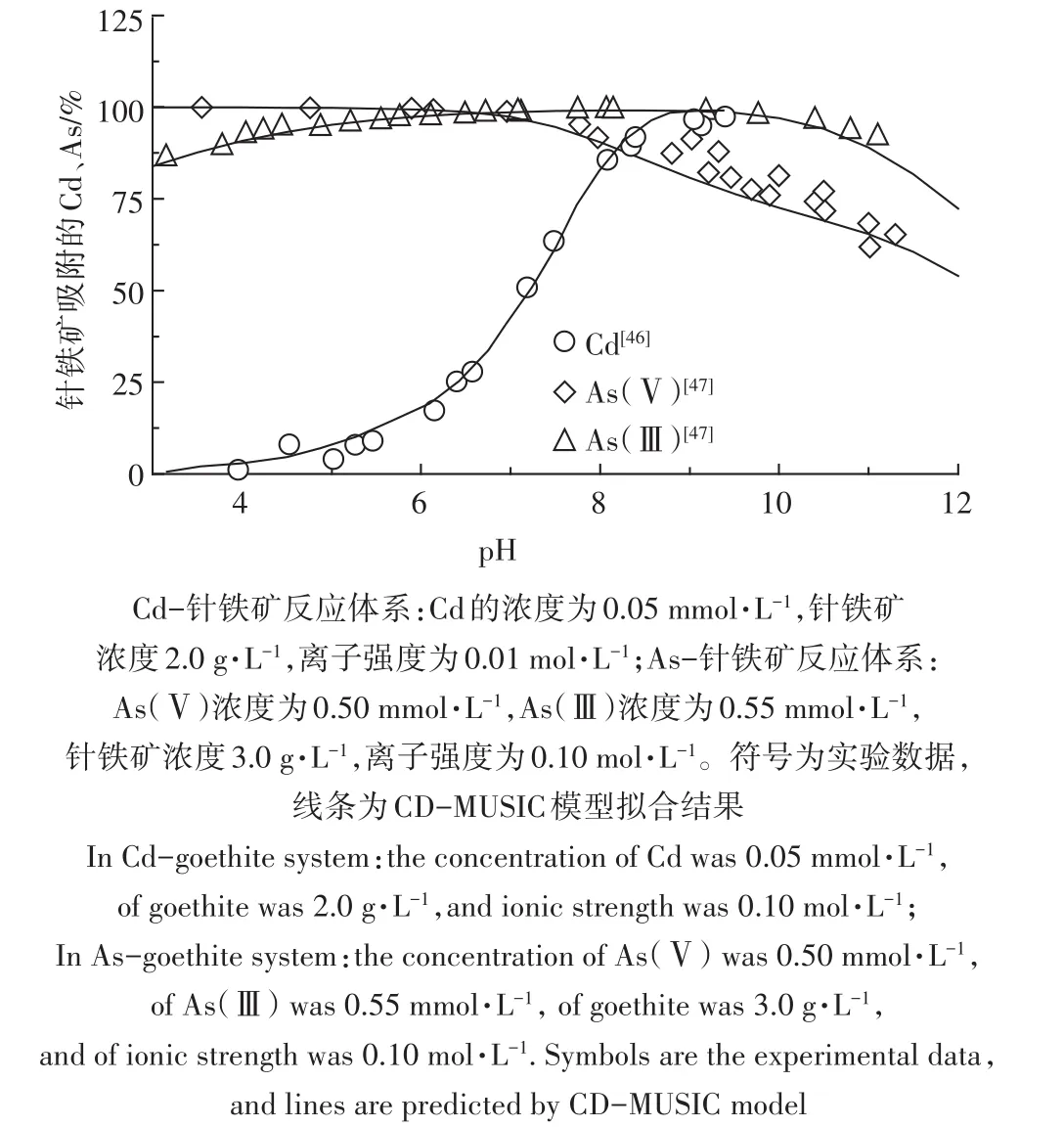
图2 镉/砷在针铁矿上的吸附[46-47]Figure 2 Adsorption of Cd/As(Ⅲ,Ⅴ)onto goethite[46-47]
下面我们对以上问题(1)和问题(2)在已有研究中的处理方式予以回顾,问题(3)将在本文的第三部分结合机理性模型在环境样品中的应用加以介绍。
2.2 吸附界面含量的确定
2.2.1 土壤有机质
金属阳离子在有机质上的吸附行为一般由分离纯化的HA和FA代表,而通常实验结果仅包括土壤有机质和可溶性有机质含量,但是有机质的组成、离子结合能力与其来源和土壤时空分布相关[57],所以在模型建立时需要确定土壤有机质离子吸附能力相当于多少HA和FA。常见的一种方法是将一定比例的有机质视为HA或者FA。如Schroder等[58]假定土壤固相体系有机质的50%为HA,土壤溶液中可溶性有机质的40%为FA,其余均为惰性有机质。第二种方法是测定土壤中HA和FA的含量,同时假定在土壤中起作用的有机质为此部分测定的HA或FA[59]。例如:Dijkstra等[60]以快速土壤腐植酸测定方法(Van Zomeren等[61])测得8种沙壤中腐植酸含量占有机质的25%~67%,Groenenberg等[62]测定的沙壤中腐植酸占有机质的81%~87%,Lofts等[63]得到FA和HA占溶解性有机质的63.5%,Ren等[64]研究得出土壤溶液中仅有26.2%的可溶性有机质为腐植酸(主要为FA)。第三种方法是由土壤总阳离子交换量(CEC)减去土壤黏土矿物的CEC得到土壤有机质的电荷密度,并由此折算成HA和FA的含量,如Weng等[65]用这种方法估量了沙壤中有机质的电荷携带量相当于含有16%~46%(平均36%)的HA。Gustafsson等[66]通过pH与溶解的Al和Ca间的浓度关系,拟合了14种土壤中腐植酸组分含量,研究结果显示土壤中活性有机质的含量在17%~84%之间。化学分析及模型拟合测定结果显示土壤中的活性有机质所占比例在14%~87%之间不等[66-67]。
2.2.2 金属氧化物
土壤中金属氧化物多以铁、铝(氢)氧化物为主,锰氧化物相对含量较少。因此,大多数计算重金属化学形态的机理模型只考虑铁或铁-铝的(氢)氧化物对重金属离子的吸附。但是,有研究指出锰氧化物对金属离子(如Pb)的吸附也有着很大贡献[68-69]。Hiemstra等[70]提出离子在铁、铝氧化物上的亲和常数十分接近,只需考虑不同氧化物比表面积的差异,离子的吸附可以由单一组分氧化物的吸附在模型中代表。铁、铝(氢)氧化物在土壤中的总含量一般采取DCB(二亚硫酸钠-柠檬酸钠-重碳酸钠)还原络合提取方法测定[71],无定形铁、铝氧化物含量由草酸铵提取确定[72]。一般将草酸铵提取的铁、铝视为无定形氧化物,而DCB与草酸铵提取量之差视为结晶态氧化物[11,73]。结晶态氧化物通常用针铁矿来代表,无定形氧化物常以水铁矿来代表[11,70]。Weng等[65]直接假设结晶态的铁、铝氧化物的比表面积为100 m2·g-1,而无定形的铁、铝氧化物的比表面积为600 m2·g-1。Hiemstra等[70]以磷酸盐在土壤上的吸附为探针,通过模型反推测了不同土壤中反应活性氧化物的比表面积。
2.2.3 黏土矿物
黏土矿物带永久性负电荷,对阳离子的吸附以静电作用为主,同时在其片状结构的侧面也有类似于金属氧化物表面的活性基团(如铝醇基)可以吸附阴离子[74]。土壤黏土颗粒(<2 μm)含量可以通过常规土壤性状分析获得,并通过矿物组成分析或对样品当地主要黏土组成的了解,以确定所研究样品中黏土矿物的主要类型,并由该类黏土矿物的CEC范围确定土壤黏土颗粒的永久性负电荷密度。Weng等[65]和Rennert等[75]的研究中确定土壤黏土矿物主要成分为伊利石,所以黏土矿物的电荷密度相当于伊利石的阳离子交换量(0.1~0.4 mol·kg-1)。在模拟含氧阴离子在黏土矿物片状结构侧面位点上的吸附时,Gustafsson[76]以水铝英石(非硅酸盐类)作为土壤黏土矿物代表,定义了吸附位点特性:铝醇基的吸附位点为4位点·nm-2。
2.3 重金属“活性”总量的确定
如上所述,强酸消解能溶解很多一般情况下难以溶解的矿物,所以采用王水消解得到的土壤重金属含量常会高估“活性”重金属(如Cd、Pb、Zn、Ni)的含量[77-78]。以2 mol·L-1硝酸提取的Cd和Cu与模型分析相符,但是该浓度硝酸会高估Ni和Zn的土壤吸附量,导致模型计算与测量值之间的差异[55],而以0.4 mol·L-1硝酸提取土壤中的Cd、Cr、Cu、Ni和Zn浓度与模型分析结果能够很好地吻合[62];也有作者使用螯合剂(如EDTA)提取土壤“活性”重金属[79-80]。对于有机土(有机质含量大于10%)、森林土和农业耕种土,以EDTA提取与以0.43 mol·L-1硝酸提取的金属含量结果基本一致[81-82]。由同位素交换反应获得的“活性”重金属含量计算得到的溶液中重金属浓度较利用王水提取的方法相比结果更为准确,尤其是对重金属Cd、Pb、Zn而言[78]。
土壤中含氧阴离子的“活性”含量常以草酸铵提取测定,其基本原理是在提取过程中随着无定形金属氧化物的溶解,吸附在金属氧化物上的含氧阴离子也释放出来,如Cui等[56]和Gustafsson[83]用草酸铵提取了土壤中的As作为“活性”As的总量。也有研究者采用酸消解/提取测定土壤“活性”含氧阴离子含量作为模型输入值总量计算离子的形态分布,但是研究发现土壤酸碱性影响模型预测与提取态离子含量拟合的结果:对于碱性土壤,采用王水消解的As模型预测结果与溶解态As测定值吻合;但在pH<6.8的土壤上,溶解态As高于模型预测值[77]。但是Dijkstra等[60]研究表明在中性及酸性条件下,使用0.43 mol·L-1硝酸提取的As能够较好地预测可溶态As,而在pH>8时,模型低估了可溶态As。对于硒、锑、钼酸盐,使用0.43 mol·L-1硝酸提取能够很好地预测溶解态硒,但所测溶解态锑与模型预测相比偏高,而所测溶解态钼偏低[60,62]。因此在模型输入值确定方面,准确选择合适的浸提剂定量分析样品中金属阳离子、含氧阴离子的浓度尤为重要。
3 机理性模型在土壤重金属化学形态计算中的应用
近二三十年,机理性化学形态模型在土壤等环境样品中的应用取得了很大的进展。其中针对金属阳离子机理性模型的发展领先于含氧阴离子。我们分别以Cd和As作为金属阳离子及含氧阴离子的代表,举例说明机理性模型在天然环境样品重金属化学形态研究中的应用。
3.1 阳离子:以Cd为例
在有氧条件下,重金属阳离子在土壤中的分配一般由吸附反应控制。土壤有机质、黏土矿物和金属氧化物均有一定的阳离子吸附能力。目前常用的方法是基于线性叠加假设的多表面模型(Multi-Surface Model)[65]。多表面模型选取纯化腐植酸、合成针铁矿等模型材料分别代表土壤中的一类吸附界面,根据这些模型材料信息建立的先进的表面络合模型及模型参数,计算离子在每种界面上的吸附量并予以加和,忽略土壤中各吸附介质的交互作用对离子吸附的影响(多表面模型示意图如图3a所示)。一系列的研究表明,多表面模型在很多情况下能够较好地预测土壤中重金属阳离子(如Cu2+、Cd2+、Zn2+、Ni2+)的化学形态分布。如:Weng等[65]用NICA-Donnan模型、CD-MUSIC模型、DDL模型和Donnan模型分别计算离子在天然有机质(固相中和可溶性有机质)、晶态和非晶态铁氧化物,以及黏土硅酸盐上的吸附,模型预测的Cd2+活度与DMT测定结果比较表明,模型能够很好地预测Cd2+在土壤中的吸附行为。同样,Cances等[84]应用相似的模型方法表明,在pH 4~6时模型预测自由态Cd2+与DMT法测定结果基本一致。Dijkstra等[85]在pH 0.4~12的范围内应用相似的模型方法研究了重金属在污染土壤中的淋溶特征,结果表明模型基本准确地预测了Cd的淋溶特征。但是,也有研究显示多表面模型在一定程度上低估了Cd在土壤中的吸附。如:Schroder等[58]研究河滩土壤(pH 5~8)金属离子的形态分布时,模型预测结果与测量结果相比基本合理,但是模型高估了大约0.5个log单位的溶解态Cd2+(图4a)。Bonten等[86]在353个非污染和污染土壤上的研究也表明,较高浓度时模型能很好地预测Cd在溶液中的浓度,而在较低浓度时模型平均高估了溶解态Cd2+。类似的报道还有Rennert等[75]的工作(图4a)。在厌氧环境下,重金属阳离子可能形成硫化物沉淀,这时模型不但要考虑吸附反应也要考虑硫化物沉淀的形成[87]。
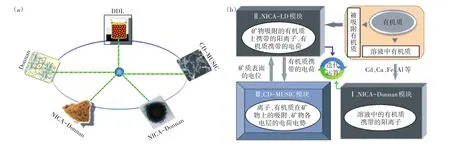
图3 多表面模型(a)和LCD模型(b)示意图(b图修改自Weng等[50])Figure 3 Schematic overview of the multi-surface model(a)and LCD model(b)(b is modified from Weng et al[50])
应用纯化胶体颗粒所得到的研究结果显示,天然有机质与氧化物等矿物表面的交互作用会影响阳离子在有机-矿物复合体系中的吸附[88-90],主要影响机制包括:静电作用、位点竞争、形成三相复合物(矿物-离子-有机质,矿物-有机质-离子)。目前的研究显示,基于线性叠加的多表面模型在许多情况下能较为可靠地预测重金属阳离子(尤其是Cu2+)在土壤中的化学形态分布,这可能是因为:(1)在土壤一般pH范围内,土壤有机质是阳离子的主要吸附界面,而土壤中与氧化物等矿物直接紧密接触的有机质只占总有机质的一部分,从而减少了有机质-矿物交互作用对离子吸附的影响;(2)虽然有机质和矿物的交互作用对离子的吸附产生了影响,但是最后的综合结果与线性叠加的结果接近,比如在FA和针铁矿的混合体系中,尽管考虑有机质-矿物交互作用的LCD模型计算指出FA和针铁矿对Cu2+吸附的贡献受到了两种吸附剂交互作用的影响,同时形成了针铁矿-Cu-FA三相复合物,但是Cu2+的吸附总量接近线性叠加结果[90]。但是对于Cd来说,如上所述,一些研究也表明多表面模型可能会低估Cd在土壤中的吸附。除了其他的不确定性以外(如模型输入值、模型参数的不确定性),天然有机质和矿物的交互作用可能会使得Cd的总吸附量高于线性叠加的结果[91]。
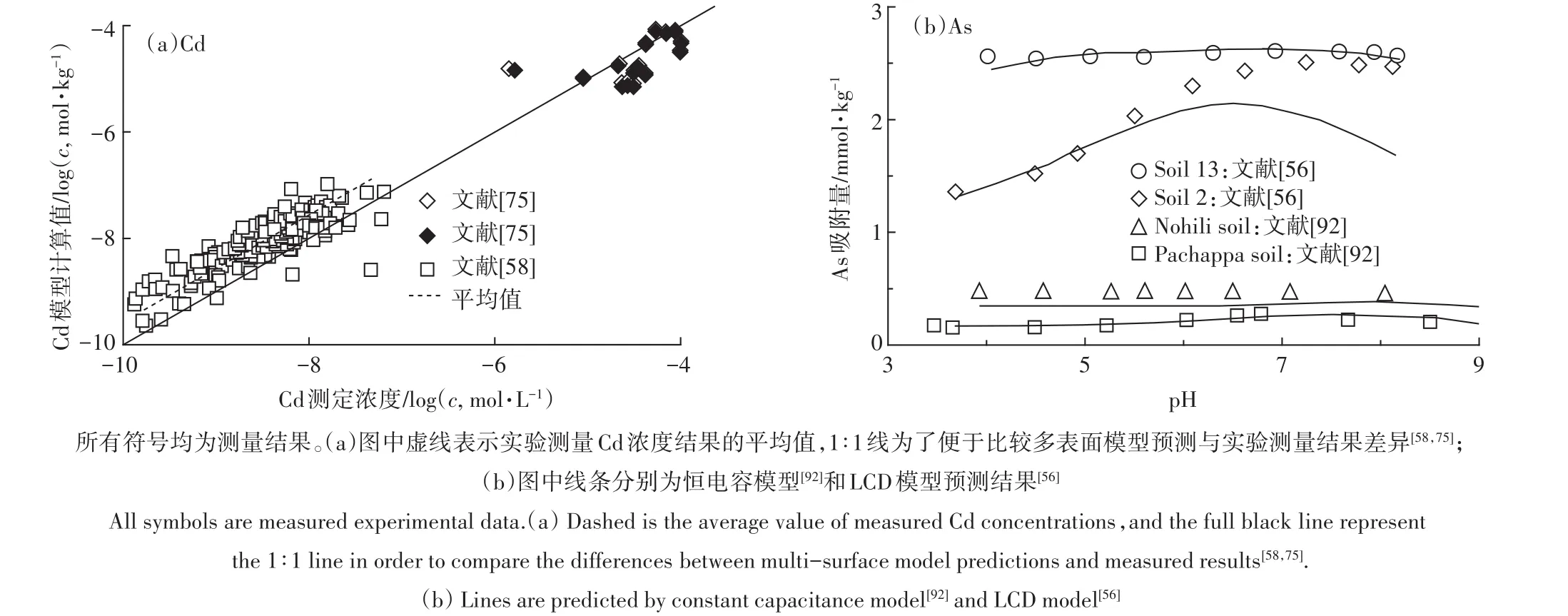
图4 镉/砷在土壤上吸附测量结果与模型预测比较Figure 4 The adsorption of Cd and As on soils by comparison with model predictions
3.2 含氧阴离子:以As为例
含氧阴离子的主要吸附界面为金属氧化物和黏土矿物。阴离子在金属氧化物上的吸附模型与阳离子在天然有机质上的模型发展基本同步,但是,当应用于天然样品时,含氧阴离子模型要落后于针对阳离子的模型,其中的一个主要原因就是如何模拟天然有机质对含氧阴离子在矿物表面的竞争吸附作用。一般表面络合模型只考虑小分子如无机离子在胶体颗粒表面的吸附,但是天然有机质是带有活性基团的大分子,它在矿物表面的吸附及对含氧阴离子的影响难以在一般表面络合模型中模拟。Gustafsson[76]应用CD-MUSIC模型研究了As(Ⅴ)在土壤上的吸附,认为土壤中的金属氧化物(水铁矿、三水铝石)和黏土矿物(水铝英石)是起主要作用的吸附界面,然而在这一模型计算中有机质在矿物上的吸附对As(Ⅴ)吸附的影响被忽略了。事实上,土壤中普遍存在的有机质对阴离子在金属氧化物等矿物表面的吸附存在着强烈的竞争作用[53,93]。在 Gustafsson[83]另一 CD-MUSIC 研究中,将有机质对As(Ⅴ)在土壤上吸附的影响简化为矿物表面基团与羧基的络合反应。与此类似,Hiemstra等[93]应用NOM-CD模型研究了腐植酸对磷在针铁矿上吸附的影响,与CD-MUSIC模型相比,Hiemstra等[93]在NOM-CD模型计算中加入了以两个羧基代表的吸附在针铁矿表面的天然有机质,并允许该羧基发生质子化反应。以羧基代表吸附在矿物表面的有机质可以将有机质对阴离子吸附的影响纳入一般表面络合模型的计算模式中,但是这一处理可能过于简化,一旦体系pH、Ca、等条件发生变化,模型就不能准确预测吸附的有机质携带电荷的变化及对含氧阴离子吸附的影响。
LCD模型结合了NICA模型和CD-MUSIC模型用于计算在天然有机质影响下土壤矿物表面发生的表面络合反应(LCD模型示意图如图3b所示)。Cui等[56]结合LCD模型研究了5种不同土壤As(Ⅴ)吸附与土壤氧化物组成的关系(其中2种土壤实验结果如图4b所示),该模型构建方法与Weng等[94]研究磷在土壤上吸附方法相似。具体来说:认为As(Ⅴ)在土壤上的吸附主要是由金属氧化物主导,以针铁矿作为土壤吸附含氧阴离子活性表面的典型代表;吸附在土壤金属氧化物上的天然有机质以分布在第一和第二Stern层的FA来代表,其中,吸附在第一Stern层的FA的羧基(RCOO-)可与针铁矿表面的单齿配体(≡FeOH)形成内圈配合物(≡FeOOCR),所有的羧基和酚羟基(RO-)可以结合H+、Ca2+、Al3+和Fe3+。吸附的有机质配体(羧基和酚羟基)与表面位点、质子及其他阳离子间的作用以NICA模型进行计算,并假定NICA模型参数和溶液中FA的参数相同。结果表明,在pH 6~8范围As(V)溶解度的降低主要是由于Ca吸附的促进作用。土壤中磷酸根的吸附强度高于As(Ⅴ),而在针铁矿上二者的吸附强度类似,这可能是由于土壤氧化物中Al替代Fe降低了对As(Ⅴ)的吸附能力[56]。天然环境中As形态受环境体系氧化还原电位的影响,在还原条件下(如:淹水水稻田),土壤中As(Ⅴ)可还原成As(Ⅲ),同时由于反应速度较慢,As(Ⅴ)和As(Ⅲ)可同时存在。因此在模型预测分析中,需要考虑不同价态的As。Cui等[56]应用HPLCICPMS测定了土壤溶液中As(Ⅴ)与As(Ⅲ)的含量,并用LCD模型计算了As(Ⅴ)和As(Ⅲ)在土壤有机-无机矿物胶体上的吸附。
3.3 计算机软件
为了增强对复杂环境样品体系中元素化学形态及迁移的计算能力,人们开发了多种计算机软件。用于水和土壤等环境样品中重金属化学形态计算的常用软件包括:Visual-MINTEQ[95]、PHREEQC[96]、ECOSAT[97]、WHAM[42]和ORCHESTRA[98]。所有这些软件的基本原理和构成大体相似,即根据化学反应平衡和质量平衡建立方程式组,由软件中的方程解锁核心通过迭代运算求出未知数值。软件中预定义了不同类型反应的数量关系,如:酸-碱反应、沉淀反应、吸附模型等,使用者可以根据所研究体系的组成特征选择相关的组分及反应类型,较为方便地去定义具体的化学反应平衡体系[98]。其中,ORCHESTRA软件采用开放模块定义某种反应类型的数量关系,用户在使用过程中可以自由地调整/增加模块,因而ORCHESTRA软件增加了使用的灵活性[99]。目前模拟天然有机质-矿物交互作用的LCD模型只在ORCHESTRA环境下运行,而基于线性叠加假设的多表面模型原则上可以在以上所有的软件环境下进行。至今CD-MUSIC和NICADonnan模型计算均可在ECOSAT、Visual-MINTEQ和ORCHESTRA中完成。
4 总结与展望
针对土壤中重金属化学形态的定量研究,模型计算可以弥补实验室分析手段的不足,获得更为全面和具有预测性的结果。近二三十年,先进的表面络合模型的发展极大地促进了机理性化学形态模型在环境样品中的应用。其中,基于线性叠加假设的多表面模型能够较为成功地预测诸如Cu、Cd、Ni、Zn等重金属阳离子在环境样品中尤其是在弱酸性条件下的化学形态分布,但目前多表面模型常低估Pb在土壤中的吸附。含氧阴离子在土壤中的化学形态模型的发展落后于针对阳离子的模型,其主要原因是线性叠加模型不能反映天然有机质对含氧阴离子在矿物表面吸附的竞争作用。近期发展起来的LCD模型是基于非线性叠加假设、涵盖有机质-矿物交互作用的表面络合模型,已经被应用到磷、砷、硒这些含氧阴离子在土壤中化学形态分布的研究中。今后的研究可以从以下几个方面入手:
(1)进一步获取与完善离子在单一吸附介质上的模型参数,如锑在金属氧化物上的CD-MUSIC模型参数。
(2)选择适合的测定“活性”重金属含量的提取剂,尤其是针对某些类型的土壤比如石灰性土壤、矿区土壤的“活性”重金属提取剂。
(3)明确适合代表土壤金属氧化物离子吸附能力的模型材料,如是否可以用实验室合成的针铁矿代表土壤中的铁氧化物。
(4)进一步验证与发展LCD模型及其在重金属离子化学形态计算中的应用。
(5)基于LCD模型运行结果,建立更为简便的有机质-矿物交互作用模型。
(6)进一步扩大重金属化学形态模型的应用范围,使其在科学研究以外获得在污染风险评估、污染环境修复领域更广泛的应用。
