1例肾上腺脊髓神经病病例报道及文献回顾*
2018-07-31林永强刘子凡许治强梁燕玲李斯颖
林永强,刘子凡△,许治强,梁燕玲,李斯颖
(广州医科大学附属第三医院神经内科,广州 510150)
X-连锁肾上腺脑白质营养不良(X-linked adrenoleukodystrophy,X-ALD)是一种脂质代谢障碍病,呈X性染色体连锁隐性遗传,在美国的患病率约为1∶ 17 000[1]。它是一种渐进的神经退行性疾病,由于体内过氧化物酶缺乏、长链脂肪酸(C23~C30)代谢障碍,极长链脂肪酸(very long chain fatty acids,VLCFAs)在体内,尤其脑和肾上腺皮质沉积,导致脑白质脱髓鞘和肾上腺皮质病变[2]。肾上腺脊髓神经病(adrenomyeloneuropethy,AMN)是X-ALD的一种变异类型[3],以进行性下肢痉挛性瘫痪、括约肌障碍和性功能障碍为主要临床表现,多在成年后发病,病情缓慢进展,预后较差,临床上对此病了解较少,容易引起误诊及治疗上的失误,早期认识此病具有重要意义。
1 资料与方法
1.1一般资料 患者,男性,29岁,因“双下肢乏力10年,加重3年”2017年8月9日入住本院神经内科。患者于10年前开始出现双下肢乏力,行走时出现右下肢拖步,伴有下肢麻木,曾服用激素治疗,3年前自行停用激素,出现双上肢乏力,四肢僵硬,近1年出现言语功能障碍,有吞咽、饮水呛咳,近半年出现颈肌无力,不能抬头。既往有肾上腺皮质功能不全病史12年。家族史无特殊,父母及哥哥均无相关临床症状。体格检查:血压正常,神清,头发、眉毛偏少,皮肤偏黑,唇部、乳晕、背部见色素沉着(图1)。言语低沉、细微,能理解问话,常规测视力正常,双眼球居中,活动无受限,向双侧凝视可见粗大眼震,双侧瞳孔等圆等大,对光反射灵敏,听力正常,软腭上提好,咽反射迟钝,鼻唇沟对称,伸舌偏左,舌肌无萎缩、震颤,颈软、无力,抬头困难,四肢肌力0级,肌张力增高,远端肌肉见萎缩,浅感觉正常,关节位置觉消失,四肢腱反射亢进,双侧Chaddocks征阳性,踝阵挛(+)。
1.2方法 入院后行相关实验室检查。2017年8月:血、尿、大便常规(-),肝肾功能、心酶、尿酸、电解质正常,凝血常规、自身免疫病组合、癌相关5项正常,人类免疫缺陷病毒、梅毒(-),甲状腺功能5项、甲炎2项正常,血清铜、血清铜蓝蛋白正常,抗内因子抗体(-),壁细胞抗体(-),寄生虫抗体(-),血糖、血酮体、血氨、血乳酸、叶酸、维生素B12水平正常,促肾上腺皮质激素435.2 pmol/L,8AM血浆皮质醇107 nmol/L,4PM血浆皮质醇197.2 nmol/L,24 h尿皮质醇60.8 nmol/L,雌二醇、促黄体生成素、促卵泡生成素、睾酮正常,泌乳素39.3 ng/mL。影像学检查:2014年外院头颅磁共振成像(MRI)未见异常(图2);脊髓MRI矢状位见脊髓后索T2高信号,横断位见颈段及上胸段脊髓轻度萎缩,后索、侧索对称性T2高信号(图3)。
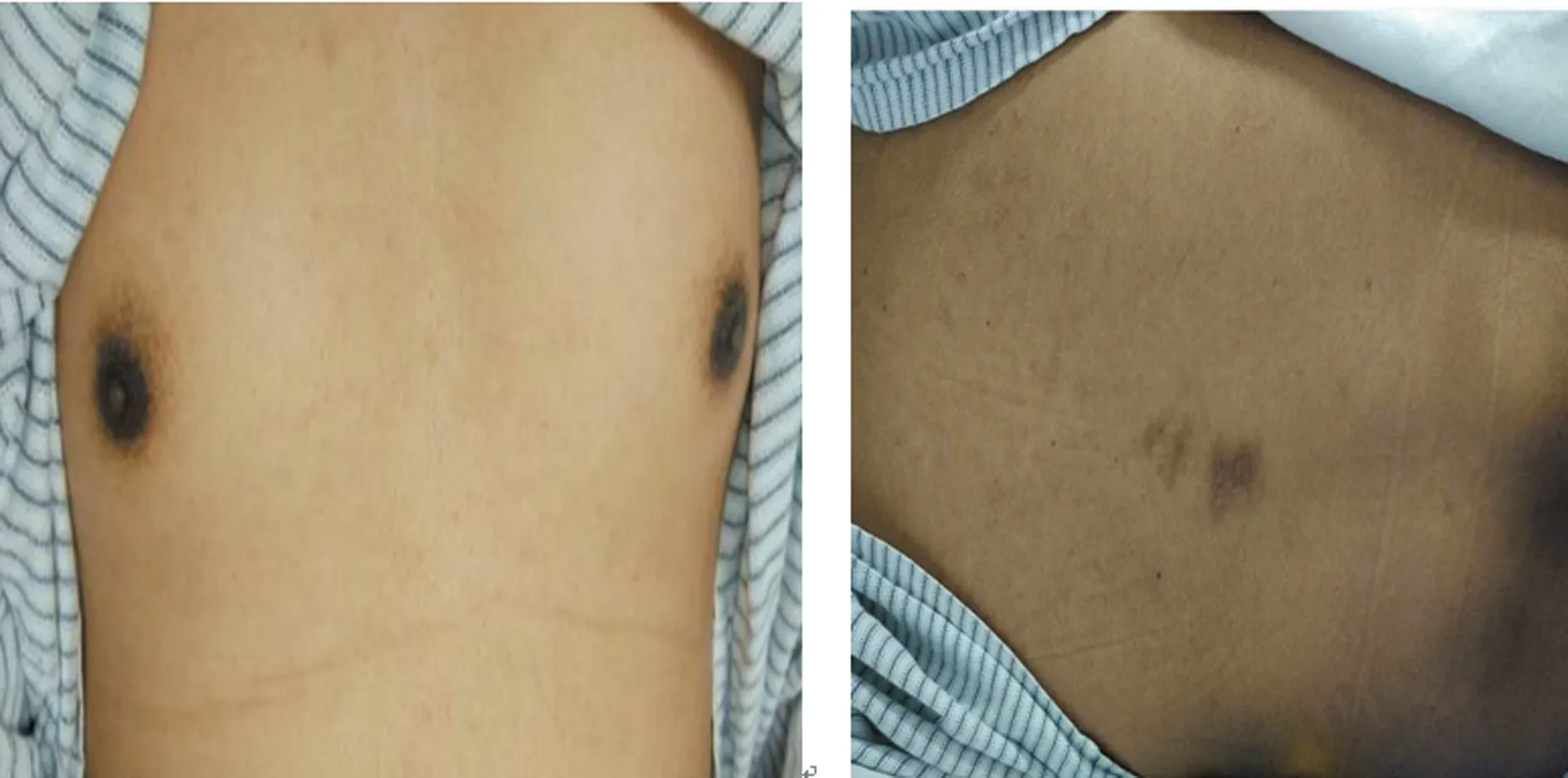
图1 体格检查照片
图2 头颅MRI(TW1)
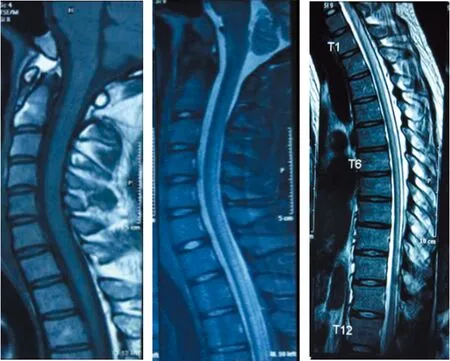
图3 脊髓MRI矢状位(脊髓后索T2高信号)
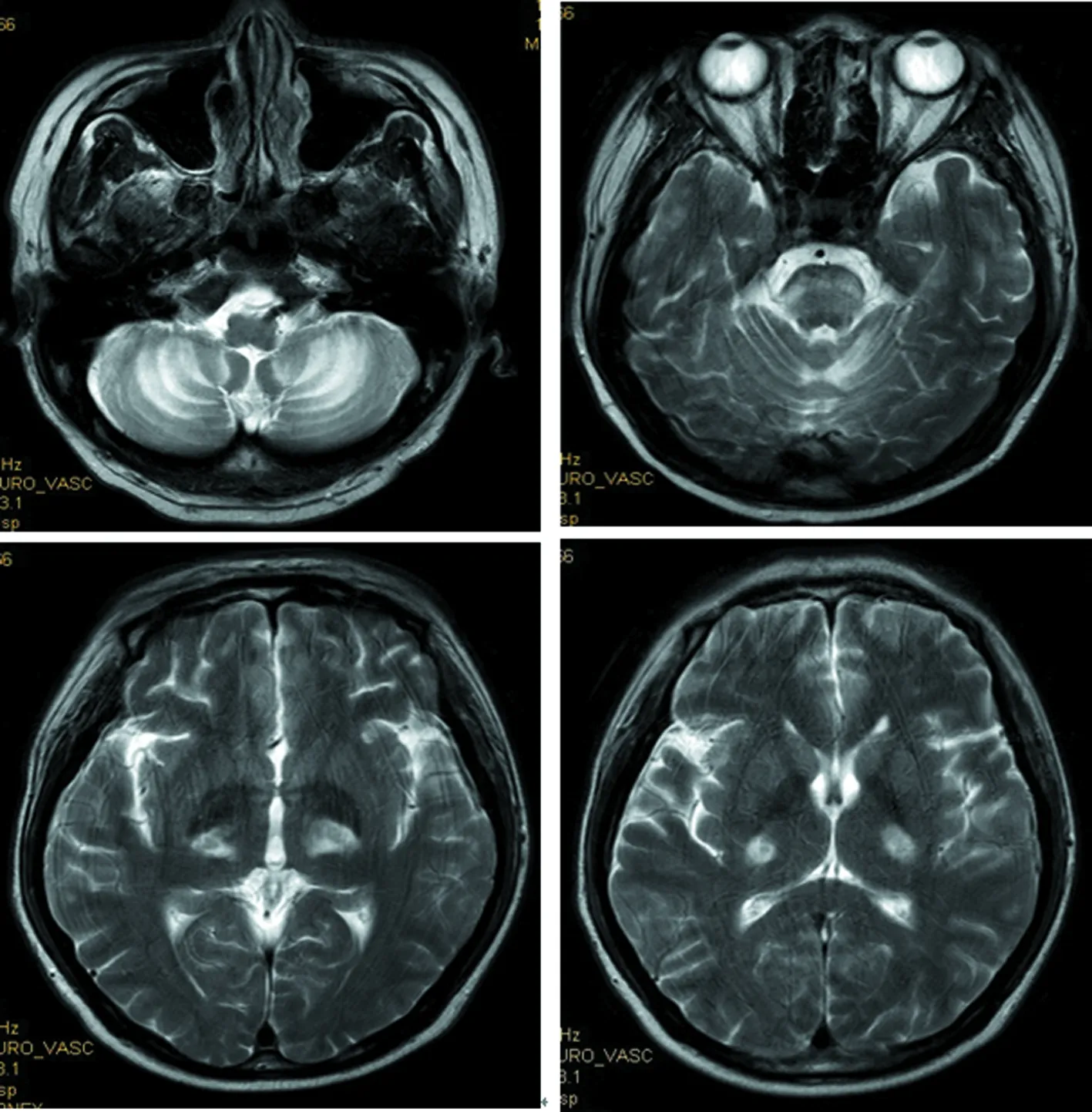
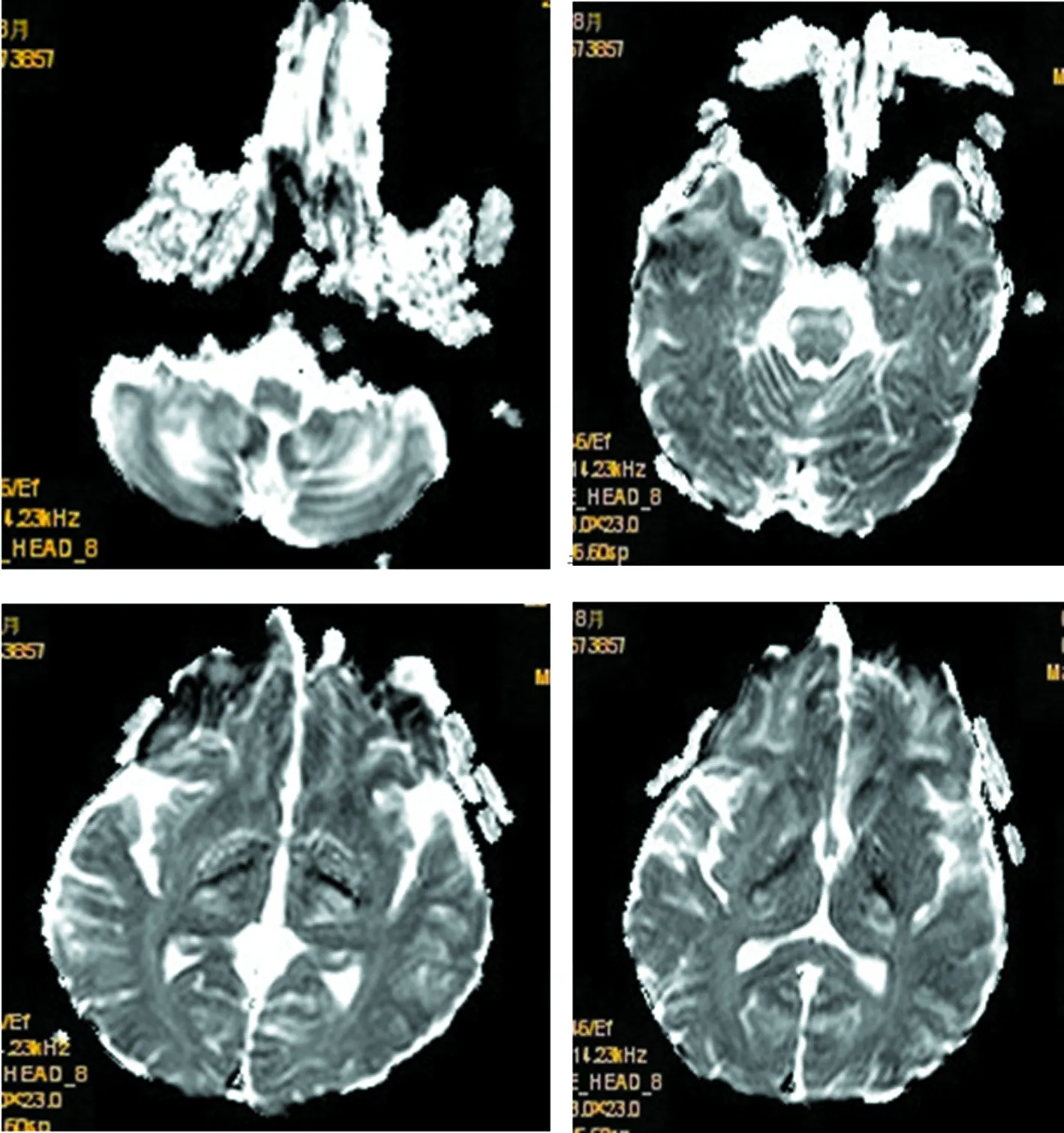
2017年8月:胸部X线片、心电图正常,肌电图:双侧正中神经感觉潜伏期延长伴传导速度减慢,双侧正中神经运动潜伏期延长,传导速度、复合肌肉动作电位(CMAP)、F波正常;头颅MRI(图4~9):双侧丘脑、大脑脚、小脑半球、脑桥、延髓见对称性长T1、长T2信号影,压水呈高信号;双侧内囊后肢病变的中央部分呈短T1、T2信号,周围部分呈长T1、T2信号。内囊后肢及小脑病变部位弥散成像、ADC图中心部分低信号,周围部分高信号,脑桥病变处弥散成像呈等信号,ADC图高信号。增强扫描:脑桥病灶部分有强化(图10)。脊髓MRI:颈段及胸段脊髓萎缩,T2成像未见异常信号(图11、12)。
颈段及上胸段后索、侧索对称性T2高信号
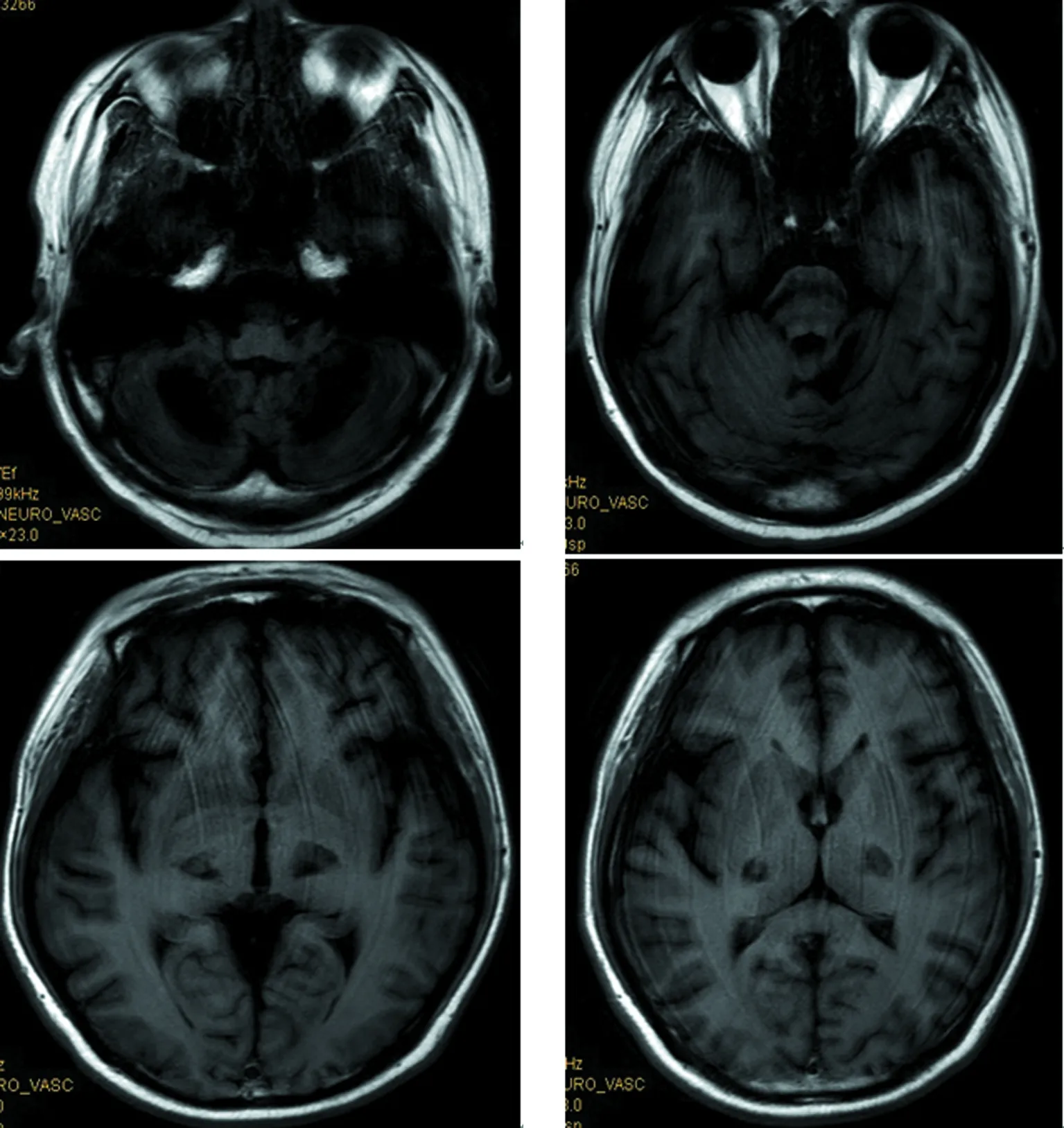
双侧丘脑、大脑脚、小脑半球、脑桥、延髓见对称性长T1信号
双侧丘脑、大脑脚、小脑半球、脑桥、延髓见对称性长T2信号
2 结 果
经科室讨论后进一步检查,血VLCFAs: C26:0 1.850 μg/mL[正常参考值范围:(0.23±0.09)μg/mL],C26:1:0.490 μg/mL[正常参考值范围:(0.18±0.09)μg/mL],Phatanic Acid:0.570 μg/mL (正常参考值范围:<0.300 μg/mL),C24:0 43.180 μg/mL[正常参考值范围:(17.59±5.36)μg/mL],C24/C22:1.886 μg/mL[正常参考值范围:(0.84±0.10)μg/mL],C26/C22:0.081 μg/mL[正常参考值范围:(0.010±0.004)μg/mL]。ABCD1基因检测(武汉康圣达医学检验所),chrX:152994732半合子变异;家系一代测序验证结果:位点为c.C946T,患者T-,父亲C-,母亲CT,哥哥T-。最后诊断:肾上腺脊髓神经病。

双侧丘脑、大脑脚、小脑半球、脑桥、延髓见对称性高信号
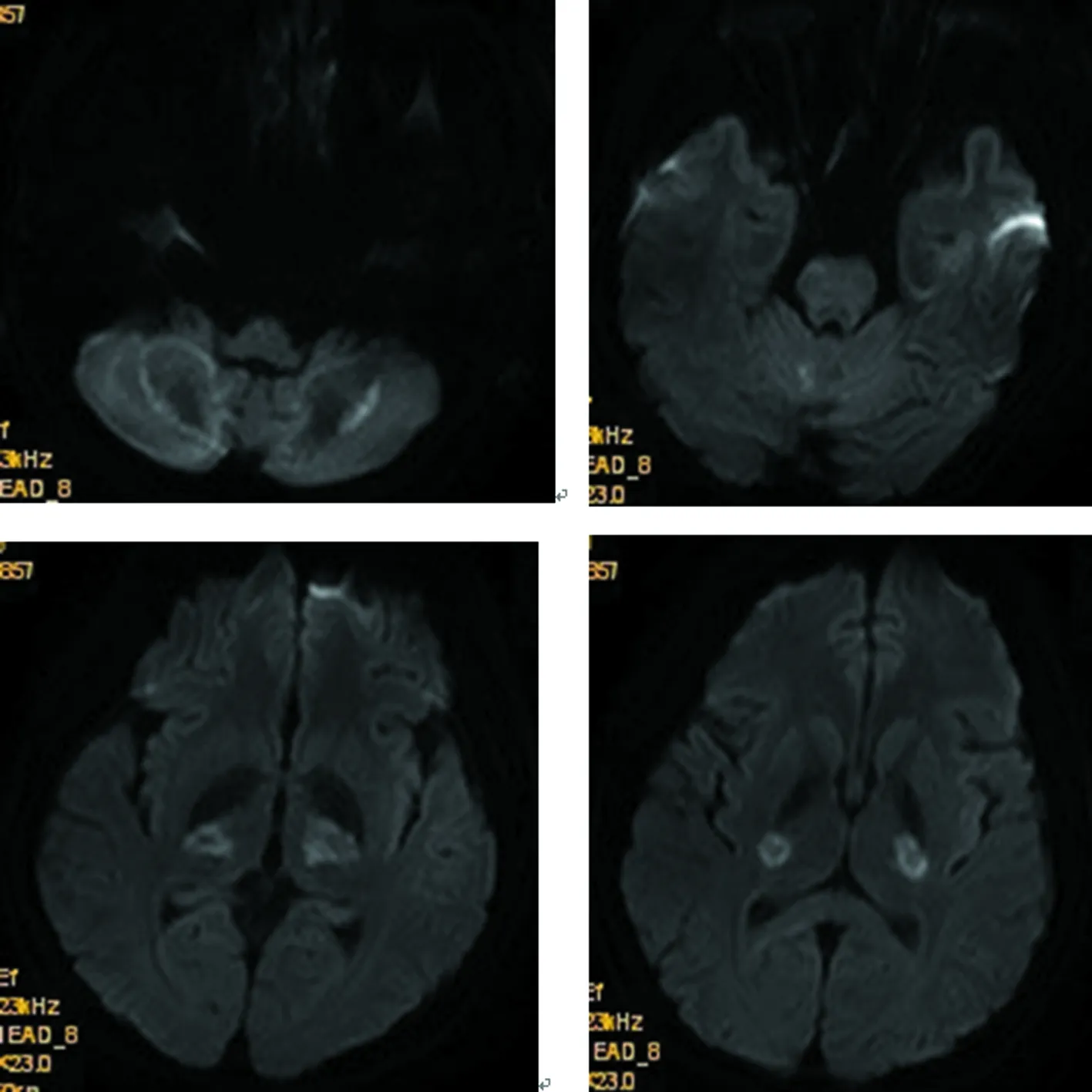
内囊后肢及小脑病变部位中心部分低信号
内囊后肢及小脑病变部位中心部分低信号
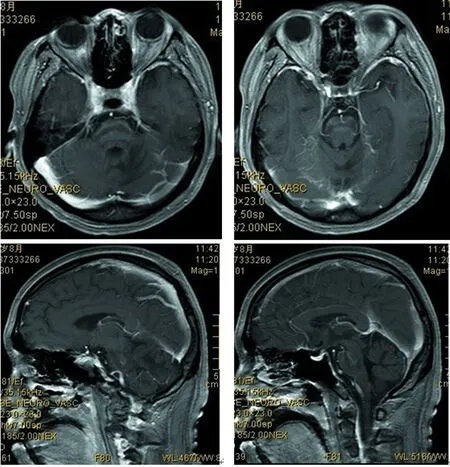
图10 头颅MRI(增强)脑桥病灶部分有强化
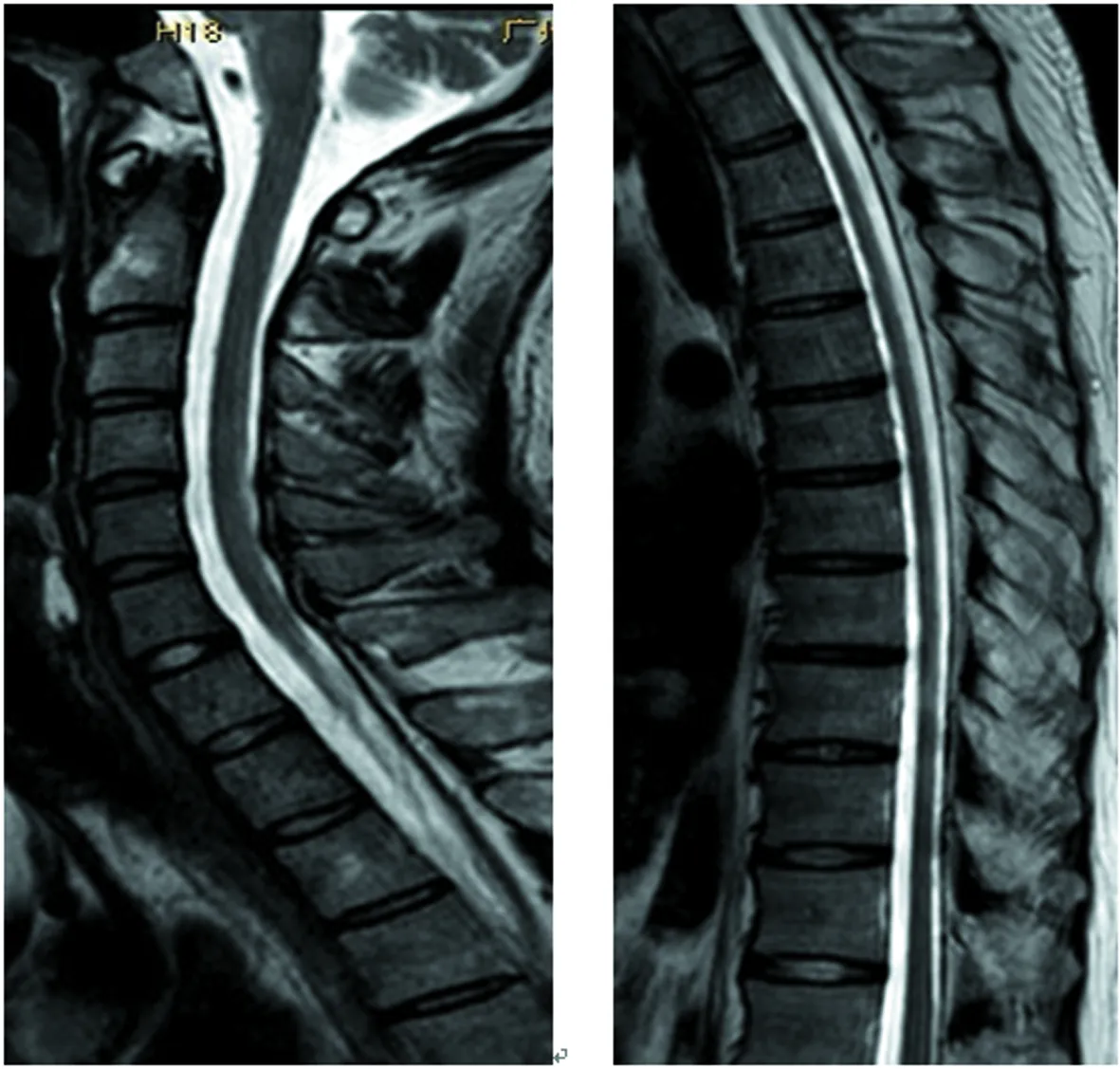
图11 脊髓MRI横矢状位(颈段及胸段脊髓萎缩)
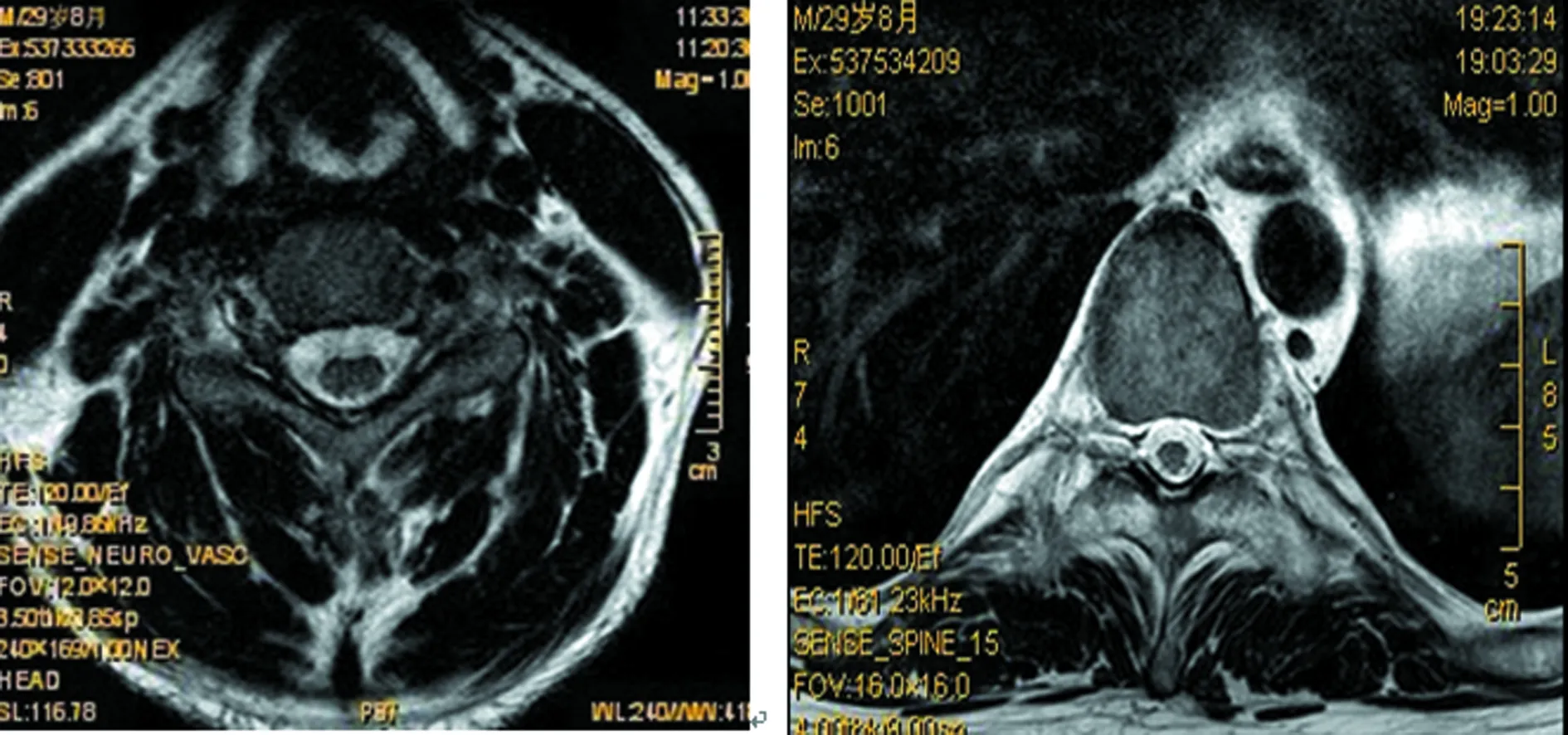
图12 脊髓MRI横断位(未见异常信号)
3 讨 论
X-ALD是ABCD1基因(Xq28)突变引起的,该基因编码ATP结合盒转运体——完整的过氧化物酶膜蛋白,参与VLCFAs(C≥22:0)的输入和长链脂肪酸辅酶A进入过氧化物酶体的降解[4]。ABCD1基因转运体的功能缺陷导致器官和组织中的VLCFAs的积累和β氧化损伤,特别是特殊的疾病标志物蜡酸(C26:0)[5],并通过几种途径影响代谢产物(前驱物或最终产物)的水平,参与VLCFAs、胆汁酸、嘌呤的代谢[6]。X-ALD及AMN的生化标志是VLCFAs(>C22:0)在各种脂质类组织中的病态积聚,主要影响中枢神经系统、肾上腺和睾丸[7]。就算在同一器官中(如大脑),不同的细胞类型,过度积聚的VLCFAs也是不同的,其临床表现差异很大[8]。
X-ALD的发病机制和病理生理是非常复杂的,基因突变对mRNA/蛋白水平的影响目前尚不完全清楚。病理机制的相关研究认为,多余的C26:0干扰线粒体氧化磷酸化功能,引起线粒体产生反应性氧化物,干扰线粒体的生物合成和钙信号[9]。尽管存在C26:0的积聚,但人类和小鼠的脊髓都没有显示细胞死亡,过量的游离VLCFAs可能诱导线粒体的去极化和细胞内钙稳态的解除[10]。这显示线粒体和过氧化物酶之间可能存在交叉对话,线粒体的二次参与可能是该疾病的罪魁祸首[11]。特定miRNA表达或功能的改变,已经证实存在于各种中枢神经系统疾病病理状态中,包括炎症、神经退行性疾病、自身免疫性疾病等[12]。miR-196a通过酶系统的调节使VLCFAs合成及超负荷,可能还参与儿童脑型ALD和AMN炎症基因的差异表达;其在儿童脑型ALD表达减少,而在AMN表达增加,其分子基础目前不明确。miR-196a的表达伴随着VLCFAs的量化,可能会作为儿童脑型ALD与AMN之间的一种产前/产后诊断的标志物[13]。
为了更好地理解X-ALD发病机制的分子机制,有学者提出了“三步”框架的假设:“第一步”:基因突变导致VLCFAs的病态积聚;“第二步”:VLCFAs诱导的表观遗传学/随机机制导致氧化性疾病;“第三步”:诱发炎症性疾病,从而产生了一个导致细胞凋亡和渐进性炎性脱髓鞘的恶性循环[14]。儿童脑型ALD星形胶质细胞炎症细胞因子的表达水平,比AMN的高,作为ABCD1基因缺乏所致的细胞特异性诱导的延伸因子极长链脂肪酸样蛋白1(ELOVL1),有助于了解在不同细胞类型中VLCFAs的负荷差别[15]。小胶质细胞被认为是突触丢失的潜在细胞介质,鉴于ABCD1基因在小胶质细胞中高度表达,与其他细胞类型密切相关的小胶质细胞功能障碍是积极参与到神经退行性变过程中的[16-17]。在人类和小鼠AMN的主要病理机制是小胶质细胞的激活,AMN的轴突变性导致皮质脊髓束选择性长度依赖性损伤[18]。在C26:0超量的情况下,神经组织中的成纤维细胞或其他细胞可被孵化繁殖,包括蛋白酶体的氧化应激或功能障碍,直接或间接依赖于ABCD1基因功能的代谢可能可以调节疾病的发作和严重程度[19]。
除了ABCD1基因突变之外,其他的遗传、表观遗传或环境因素也可能与X-ALD临床表现相关[20]。儿童脑型ALD和AMN的病理学基础是ABCD1基因的突变或缺乏。VLCFAs在不同的细胞类型中积聚程度不同,会产生不同程度的氧化和炎症,从而影响到中枢神经系统不同的部位和出现不同的表型[8]。X-ALD中最严重的是儿童脑型ALD,表现为儿童期大脑急性脱髓鞘,进展迅速,淋巴细胞明显浸润,在围绕炎性病变周围有明显的小胶质细胞死亡区域[21]。AMN是影响到成年人的晚发型X-ALD,通常在20~50岁,有周围神经病变和远端轴突及脊髓的皮质脊髓束受累,如本例患者表现为进行性痉挛性瘫痪为主要症状及脊髓MRI信号改变,无明显的脑部炎症或脱髓鞘[22]。约20%的AMN患者在以后的病程中有大脑脱髓鞘的表现,这种风险与早发型有关,通常在20~35岁,45岁以后其发病率下降[23]。
除了儿童脑型ALD和AMN这两种主要的表型,Addison型、脊髓脑桥小脑型和无症状型也有偶发报道,这种临床表现的异质性使X-ALD早期难以诊断[24]。排尿障碍、便秘、多发性神经病和Addison′s病可作为诊断的线索,在疾病发展过程中,70%的AMN患者有肾上腺功能不全,20%有脑部病变,所以正确的诊断是至关重要的。与其他先天性代谢缺陷疾病相一致,脑部病灶通常也是对称的[22]。如本例患者表现为皮肤偏黑,唇部、乳晕、背部见色素沉着,双侧内囊MRI有信号改变。
AMN患者应该检测血浆VLCFAs水平,在VLCFAs检测结果不明确的情况下要进行基因检测[25]。本例患者ABCD1基因在chrX:152994732位置上发生碱基C>T的半合子变异,该变异为无义突变,导致编码的氨基酸终止翻译,受检者ABCD1基因c.C946T位点变异来源于其母亲基因组,其母亲在该位点为杂合变异,其父亲在该位点为野生型,其哥哥在该位点为半合子变异,由于其父母及哥哥均无相似临床症状,故推测ABCD1基因c.C946T位点变异可能不是受检者疾病的致病位点。但有时基因检测也不能明确诊断,其原因可能是患者有一个或多个外显子大量缺失,所以不能用直接测序法确定,或者由于ABCD1基因在其他同源染色体上存在,以致于基因突变分析未能识别ABCD1基因的变化,可通过重复试验和改变引物来进一步证实[26]。VLCFAs病态的积聚可作为X-ALD的诊断试验,但它不能区分X-ALD的表型[1]。在12例AMN患者的分析中显示,突变分析显示其中10例有不同的ABCD1基因突变,有1例是新突变,所有患者脊髓MRI均显示弥漫性脊髓萎缩或微小信号,4例神经传导检查异常,3例视觉诱发试验异常,所有患者体感诱发电位显示有中枢传导障碍[27]。1例通过检测VLCFAs浓度确诊的33岁男性AMN患者,表现为双侧进行性痉挛性截瘫、阳痿和急迫性尿失禁,伴原发性肾上腺功能衰竭,其胫后神经的电生理检查发现,唯一异常的是体感诱发电位,有必要进行电生理学的随访[28]。
AMN患者脊髓MRI通常显示的非特异性脊髓萎缩和在皮质脊髓束的病变,有可能是Wallerian变性[22]。有些AMN患者在脑干锥体束MRI显示有高度不对称的信号减弱和单侧T2加权高信号[29]。联系复杂的炎症网络的糖脂和甘油干扰信号,可能是AMN潜在的疾病修改节点,为量身定制的治疗药物的发展和生物标志物的鉴定提供新机遇。某些甲基衍生物的积聚可能有潜在的致病作用,表明ABCD1转运体可能参与这些化合物的降解,而这些化合物可能作为AMN潜在的生物标志物[19]。
该疾病目前无特异性的治疗。当有肾上腺功能不足的临床表现时,可选用糖皮质激素替代治疗,本例患者曾予激素治疗,但未能阻止病情进展。或者给予患者饮食疗法治疗,Lorenzo′s油含有约20%的芥酸和80%的油酸,芥酸可能会延缓疾病早期神经系统症状的进展[30]。但也有研究表明,Lorenzo′s油可使血浆VLCFAs水平在1个月内恢复正常。在早期阶段,Lorenzo′s油治疗可以使VLCFAs的清除得到改善和恢复ACTH受体的正常活性,使肾上腺功能减退有潜在逆转的可能[31]。由于芥酸可引起动物的心脏毒性,尽管其对人类心脏毒性尚未得到证实,但高芥酸含量的油是不鼓励使用的[32]。血浆中VLCFAs水平与表型或疾病严重程度并无相关性,而降低VLCFAs的治疗尝试迄今为止对AMN的进展没有影响。
也有研究尝试用他汀类药物治疗,其短期的临床益处是减少强直和自发性的肌肉痉挛,但其不能阻止疾病的发展[33]。炎性是严重的儿童中枢神经系统脱髓鞘及脊髓和周围神经变性的一部分,静脉注射免疫球蛋白对严重的下肢疼痛可起到较好的治疗效果,可作为AMN患者伴有炎性所致顽固性疼痛的治疗手段[34]。利用慢病毒载体的基因治疗方法去纠正CD34+细胞的ABCD1基因cDNA,可能是一个很好的微创替代移植方法[35]。在康复训练方面,瑜伽治疗改善了患者的髋部、膝部和脚踝的屈曲,改善了患者的行走和在没有辅助装置的情况下站立和平衡的能力,其概念和方法为临床提供了一种低风险、低成本的治疗方法[36]。
X-ALD患者临床表型上的不同,可有不同的预后,伴中枢神经系统炎症性疾病死于儿童早期,而没有炎性疾病的周围神经系统疾病患者可活到50岁或60岁[13]。约35%的AMN患者随后发展为中枢神经系统炎性脱髓鞘,和儿童脑型ALD患者一样预后不良[37]。有研究发现,X-ALD/AMN患者血清睾酮水平均在正常范围内的水平,但42.9%的患者血清促黄体生成素和促卵泡生成素水平是升高的。患者的杂合子女儿血清VLCFAs水平升高,但没有发现其生育能力显著降低[14]。
尽管AMN在临床中少见,但对成年人以进行性下肢痉挛性瘫痪、括约肌障碍和性功能障碍为主要临床表现,特别是伴有肾上腺功能减退的患者,应该进行血浆VLCFAs水平的检测,必要时行基因检测进一步明确诊断,减少误诊及漏诊。但本病目前无特效治疗方法,激素替代疗法及降低VLCFAs治疗并不能阻止病情进展,基因治疗等其他治疗方式的疗效有待于进一步证实。
