水稻秸秆堆肥发酵粗制肥料中微生物多样性研究
2018-07-30曾椿淋施爱平毛罕平
朱 琳 曾椿淋 高 凤 施爱平 毛罕平 魏 巍
(1.江苏大学食品与生物工程学院,镇江 212013; 2.江苏大学农业装备工程学院,镇江 212013)
0 引言
农作物秸秆具有丰富的氮、磷、钾及有机质养分,是我国传统的有机肥料源[1-2],但需通过微生物发酵腐熟后才适合大田使用[3-4]。农作物秸秆腐熟过程是微生物在好氧或厌氧条件下,将秸秆中有机物质分解成为CO2、H2O、NH3以及腐殖质的过程[5]。发酵腐熟农作物秸秆的微生物种类繁多,包括细菌和多种类型的真核微生物[5-7]。这些微生物在秸秆发酵过程中进行迅速的群落结构演替,完成秸秆的腐熟过程[8]。同时,秸秆中的碳、氮含量也影响着腐解微生物的生长代谢过程[5]。作物秸秆碳氮比过高会导致微生物分解速度降低、堆肥升温缓慢、成肥周期长[7]。有研究表明秸秆中碳氮比控制在25~30较为适合微生物发酵腐熟[9]。水稻秸秆碳氮比在60~70之间,因此其堆肥发酵前必须补充适量的氮源以降低碳氮比。尿素和粪肥是秸秆堆肥发酵时常用的补充氮源。其中,尿素作为无机氮源成分单一且稳定,可直接被化能自养微生物代谢利用;粪肥作为有机氮源营养丰富,富含蛋白质、游离氨基酸、糖类等,可促使化能异养微生物生长旺盛。由此可见,秸秆发酵前氮源补充的种类不同可能会引起腐解微生物群落的结构差异,进而造成秸秆堆肥发酵过程和质量的差异。因此,有必要对经不同氮源补充制得的秸秆粗制肥料中微生物群落进行解析。
近年来,研究者已采用末端限制性片段长度多态性分析(Terminal restriction fragment length polymorphism,T-RFLP)、聚合酶链式反应-变性梯度凝胶电泳(Polymerase chain reaction-denaturing gradient gel electrophoresis,PCR-DGGE)、克隆文库等分子方法分析了发酵堆肥内微生物群落[10-12],但仍难以全面监测多样的微生物类群[13],使解析农作物秸秆腐解过程中微生物类群结构演替过程受到极大的限制。新一代基于Illumina平台的高通量测序技术的出现,可以更全面和准确地描述秸秆腐熟堆肥内微生物群落信息,为探究秸秆腐熟堆肥中微生物群落构成及功能提供了有效手段[14]。本文运用高通量测序技术对添加无机氮源(尿素)和有机氮源(牛粪)的水稻秸秆,经堆肥发酵后制得粗制肥料中细菌和真核微生物(真菌、藻类和原生动物)的群落结构多样性和差异进行研究,为进一步揭示水稻秸秆腐熟堆肥中微生物群落演替及其功能性提供数据基础。
1 材料与方法
1.1 秸秆堆肥
水稻稻秆来源于江苏省镇江市新区水稻田,新鲜无霉变,切成长度2~3 cm的秸秆段备用。为了探讨不同氮源添加对水稻秸秆发酵制得粗制肥料中微生物群落结构的影响,试验设计了2种等氮素含量的秸秆物料发酵处理,分别为:处理1,秸秆段(140 kg)+尿素(2.9 kg);处理2,秸秆段(140 kg)+干牛粪(12.1 kg)。尿素等为市售肥料。牛粪取自江苏省镇江市郊区牛场的新鲜粪肥(含水率为85%),经过高压灭菌去除自带微生物对堆肥发酵作用的影响,再经干燥备用。供试水稻秸秆、尿素和牛粪经干燥后用球磨仪粉碎,应用Vario MAX 型碳氮元素分析仪对其碳氮含量进行测定,并计算碳氮比。水稻稻秆碳氮质量分数分别为42%和0.6%(碳氮比为70),尿素碳氮质量分数分别为18%和42%(碳氮比为0.4),干牛粪碳氮质量分数分别为75%和10%(碳氮比为7.5)。
在堆肥场地制备两处水稻秸秆物料堆,尺寸约1.5 m×1.5 m×1.5 m。每铺垫约30 cm厚的秸秆段添加一次氮源。其中,牛粪均匀加入到物料表面并洒水,尿素则溶于水后均匀洒于物料表面。物料最终含水率控制在60%。物料堆制作完毕后覆盖塑料膜,自然发酵30 d后,形成粗制有机肥供后续试验。经处理1和处理2得到的粗制有机肥pH值分别为7.9和8.0。应用浓酸水解法测量[15]处理1和处理2的纤维素质量分数分别为1.2%和1.4%,半纤维素质量分数分别为6.2%和5.9%,木质素质量分数分别为22.7%和25.6%。
1.2 样品采集和DNA提取
本试验没有关注堆肥温度及腐熟时间对微生物的影响,仅对堆肥制备得到的粗制肥料中微生物群落进行初步研究。因此应用自制的取样器,分别对每个处理得到的粗制肥料进行6个采样点(包括堆肥的内部和外部各3个位点)的随机取样,再将6个采样点的样品充分混合成一份,以其中的微生物来代表该粗制肥料所有微生物类群。采用大剂量DNA提取试剂盒进行微生物基因组DNA的提取。取5 g混合后的粗制肥料,应用日本和光公司的DNA提取试剂盒Large ISOIL Beads Beating,按照操作说明进行微生物基因组总DNA的提取。提取后的DNA应用Nanodrop one 进行浓度的测定。两样品DNA质量浓度分别为1 330 ng/μL和1 450 ng/μL。
1.3 PCR扩增及高通量测序
本试验采用细菌16S rRNA和真核微生物18S rRNA的V4区域作为目标DNA序列进行PCR(聚合酶链式反应)扩增。以通用引物515F(5′-GTGCCAGCMGCCGCGGTAA-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′),并各自添加不同的条形码标记(Barcode序列)后对2个处理粗制肥料中细菌16S rRNA的V4区域[16]进行扩增。以通用引物528F(5′-GCGGTAATTCCAGCTCCAA-3′)和706R(5′-AATCCRAGAATTTCACCTCT-3′),并各自条形码标记后对2个处理粗制肥料中真核微生物18S rRNA 的V4区域[17]进行扩增。
PCR扩增均采用Takara试剂公司的HSTaq酶进行,反应条件均为:95℃预变性5 min;95℃变性30 s,55℃退火30 s,72℃延伸40 s,25个循环;72℃延伸10 min。扩增结束后,PCR扩增产物使用2%琼脂糖进行凝胶电泳,检查扩增效果。将样品的PCR产物送至北京诺禾至源科技有限公司,在Illumina-HiSeq平台上进行高通量测序。
1.4 数据分析处理
对高通量测序初始数据进行质量控制,以获得更为精准、高质量的DNA序列信息,采用Mothur 软件将得到的16S rDNA基因序列在RDP(Ribosomal database project)数据库中、18S rDNA基因序列在Silva数据库中进行嵌合体检验,充分去除嵌合体序列。为了得到每个OTU(操作分类单元)对应的物种分类信息,采用RDP classifier 贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析,用Mothur 软件构建稀释性曲线[18-19]。
利用QIIME 软件计算样品Chao1丰富度指数以及Shannon多样性指数和 Simpson多样性指数[19]。其中群落丰富度指数,其值越高表明群落物种的丰富度越高;而多样性指数可以反映样品的多样性程度,其值越大表明样品群落多样性越高。
2 结果与分析
2.1 两种粗制肥料中细菌和真核微生物群落多样性
通过对细菌16S rRNA的V4区测序,加入尿素和牛粪的两个粗制肥料处理分别得到原始序列52 949条和46 124条,经去除低质量、条形码标记(Barcode序列)和引物序列后,2个样品分别得到52 535条和45 748条有效序列,分别获得897个和954个细菌OTU(表1)。
通过对真核微生物18S rRNA 的V4区测序,加入尿素和牛粪的两个粗制肥料处理分别得到原始序列56 396条和75 399条,经去除低质量、条形码标记和引物序列后,2个粗制肥料处理分别得到55 713条和74 116条有效序列,分别获得508个和585个真核微生物OTU(表1)。
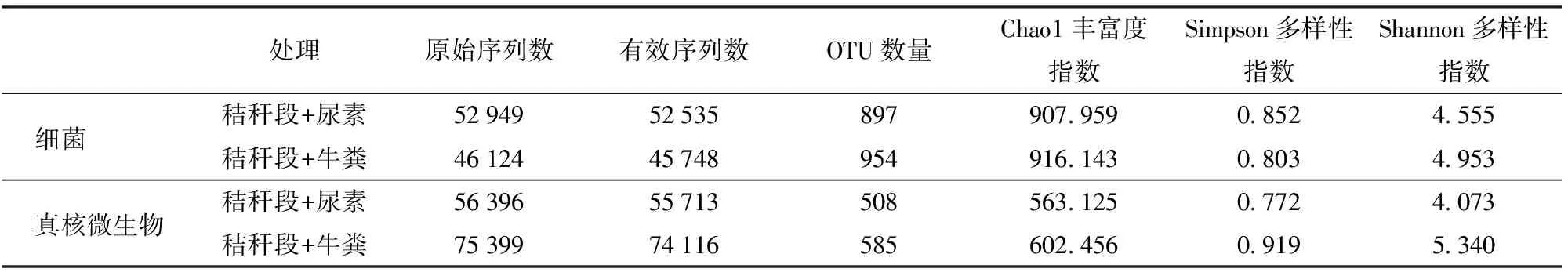
表1 两种粗制肥料的高通量测序文库质量汇总Tab.1 Information of high-throughput DNA sequencing library of two treatments
根据细菌16S rRNA测序稀释性曲线(图1a),当测序序列数量超过40 000条时,虽然仍有新的OTU 被发现,但是整个曲线已经趋于平缓,说明更深的测序几乎不会产生更多的OTU,该测序文库已经达到饱和。从图中也可以看出相同序列数时,添加牛粪的处理细菌群落OTU 多于添加尿素的堆肥处理,说明添加牛粪造成的细菌群落丰富度高于添加尿素处理。此外,通过比较2个处理的Chao1丰富度指数和Shannon多样性指数同样可以发现,添加牛粪堆肥处理的细菌群落的丰富度和多样性均高于添加尿素的堆肥处理(表1)。
根据真核微生物18S rRNA测序的稀释性曲线(图1b),当测序序列数量超过50 000条时,整个曲线已经趋于平缓,说明更深的测序不会产生更多的OTU,该测序文库已经达到饱和。与细菌测序结果一样,添加牛粪的处理真核微生物群落OTU多于添加尿素的堆肥处理,说明添加牛粪造成的真核微生物群落丰富度高于添加尿素的处理。此外,通过比较2个处理的Chao1丰富度指数、Simpson多样性指数和Shannon多样性指数同样可以发现,添加牛粪的堆肥处理真核微生物群落的丰富度和多样性均高于添加尿素的堆肥处理(表1)。
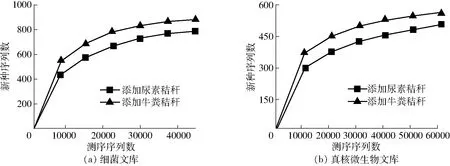
图1 细菌和真核微生物群落高通量测序文库稀释性曲线Fig.1 Rarefaction curves of high-throughput DNA sequencing library of bacterial and eukaryotic microbial community
2.2 两种粗制肥料中细菌群落组成
两个样品细菌群落在门和属分类水平上的相对丰度结果如图2所示。在门的分类水平上,两个样品中细菌群落结构组成情况相似,但各细菌类群所占比例有较大差异。如图2a所示,两个处理中细菌群落主要隶属于4个门(比例大于总序列量的1%),其中放线菌门(Actinobacteria,71.9%)在尿素处理中占主要优势,随后是厚壁菌门(Firmicutes,20.9%)、变形菌门(Proteobacteria,3.7%)、拟杆菌门(Bacteroidetes,1.7%)。而在牛粪中,变形菌门(Proteobacteria,58.5%)占主要优势,随后是厚壁菌门(Firmicutes,20.0%)、拟杆菌门(Bacteroidetes,15.8%)、放线菌门(Actinobacteria,2.8%)。
在属的分类水平上,两个样品中细菌群落结构组成和主要优势属所占比例均有较大差异(图2b)。其中尿素处理中占优势的细菌菌属分别为Streptomyces(放线菌属,40.9%)、Cellulosimicrobium(纤维菌属,22.2%)、Pediococcus(小球菌属,3.9%)、Paenibacillus(类芽孢杆菌属,3.2%)、Lactobacillus(乳酸菌属,2.7%)、Nonomuraea(野野村菌属,2.0%)。牛粪处理中占优势的细菌菌属分别为Pseudomonas(假单胞杆菌属,47.8%)、Carnobacterium(肉食杆菌属,3.6%)、Mobilitalea(无中文名称,3.0%)、Marinospirillum(海螺菌属,2.2%)。
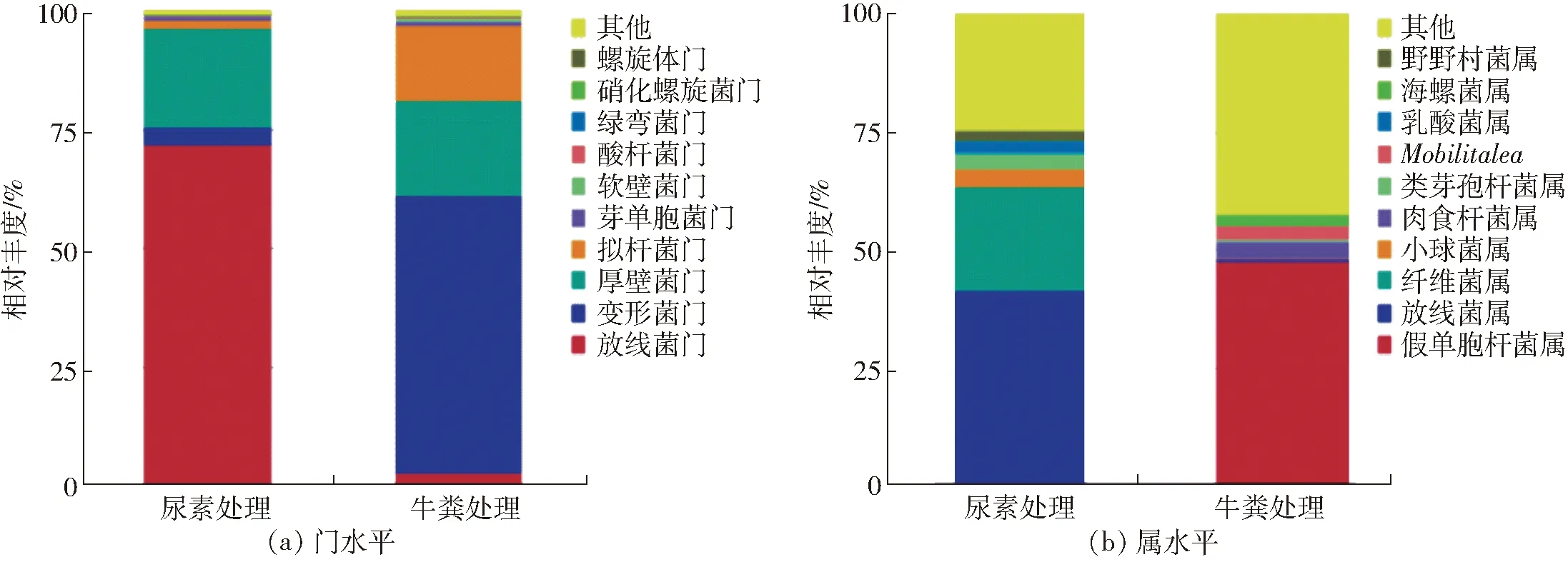
图2 两种粗制肥料中细菌在门和属分类水平的TOP10群落结构Fig.2 Composition of bacterial microbial community at phylum and genus level in two treatments
2.3 两种粗制肥料中真核微生物群落组成
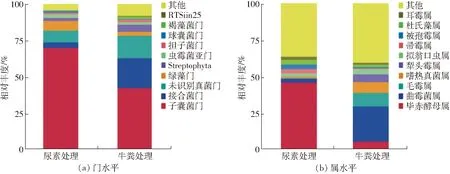
图3 两种粗制肥料中真核微生物在门和属分类水平的群落结构Fig.3 Composition eukaryotic community at phylum and genus level in two treatments
两个样品真核微生物群落在门和属分类水平上的相对丰度结果如图3所示。在门的分类水平上,两个处理中真核生物群落主要隶属于7个门,其中真菌均占据优势地位(图3a)。子囊菌门(Ascomycota,70.0%)在尿素处理中占主要优势,随后是绿藻门(Chlorophyta,6.6%)、接合菌门(Zygomycota,3.8%)、虫霉菌亚门(Entomophthoromycotina,2.3%)、球囊菌门(Glomeromycota,1.4%)。而在牛粪处理中,子囊菌门(Ascomycota,42.5%)仍占据优势,随后是接合菌门(Zygomycota,20.5%)、绿藻门(Chlorophyta,2.7%)、担子菌门(Basidiomycota,2.1%)、褐藻菌门(Ochrophyta,1.3%)、虫霉菌亚门(Entomophthoromycotina,1.3%)、球囊菌门(Glomeromycota,1.0%)。
在属的分类水平上,如图3b所示,两个样品中真核微生物群落结构组成较为相似,均以真菌菌属为绝对优势,但主要优势属所占比例均有较大差异。其中尿素处理中占优势的真核微生物属分别为Pichia(毕赤酵母属,46.1%)、Scopulariopsis(帚霉属,3.1%)、Mortierella(被孢霉属,3.1%)、Dunaliella(杜氏藻属,2.9%)、Aspergillus(曲霉菌属,2.5% )、Conidiobolus(耳霉属,2.3%)、Thermomyces(嗜热真菌属,1.1%)。牛粪处理中占优势的真核微生物属分别为Aspergillus( 曲霉菌属,23.9% )、Mucor(毛霉属,9.4%)、Thermomyces(嗜热真菌属,7.4%)、Pichia(毕赤酵母属,5.6%)、Absidia(犁头霉属,5.4%)、Dunaliella(杜氏藻属,1.7%)、Conidiobolus(耳霉属,1.3%)。
2.4 不同处理微生物群落相似性分析
通过维恩图可以直观地表现两种堆肥处理的细菌和真核微生物群落的OTU 数目组成相似性、重叠情况以及特异性(图4)。添加尿素和牛粪制得的两种粗制肥料之间共有的细菌OTU 数量为689(图4a)。其中,添加牛粪制得的粗制肥料中特异性细菌OTU数量为219,是添加尿素制得肥料中的1.7倍。同时,两种粗制肥料之间共有的真核微生物OTU为416(图4b)。其中,添加牛粪制得的粗制肥料中特异性真核微生物OTU数量为155,同样是添加尿素制得肥料中的1.7倍。因此,添加牛粪制得的粗制肥料较添加尿素制得的肥料具有更多的特异性细菌和真核微生物生物类群。
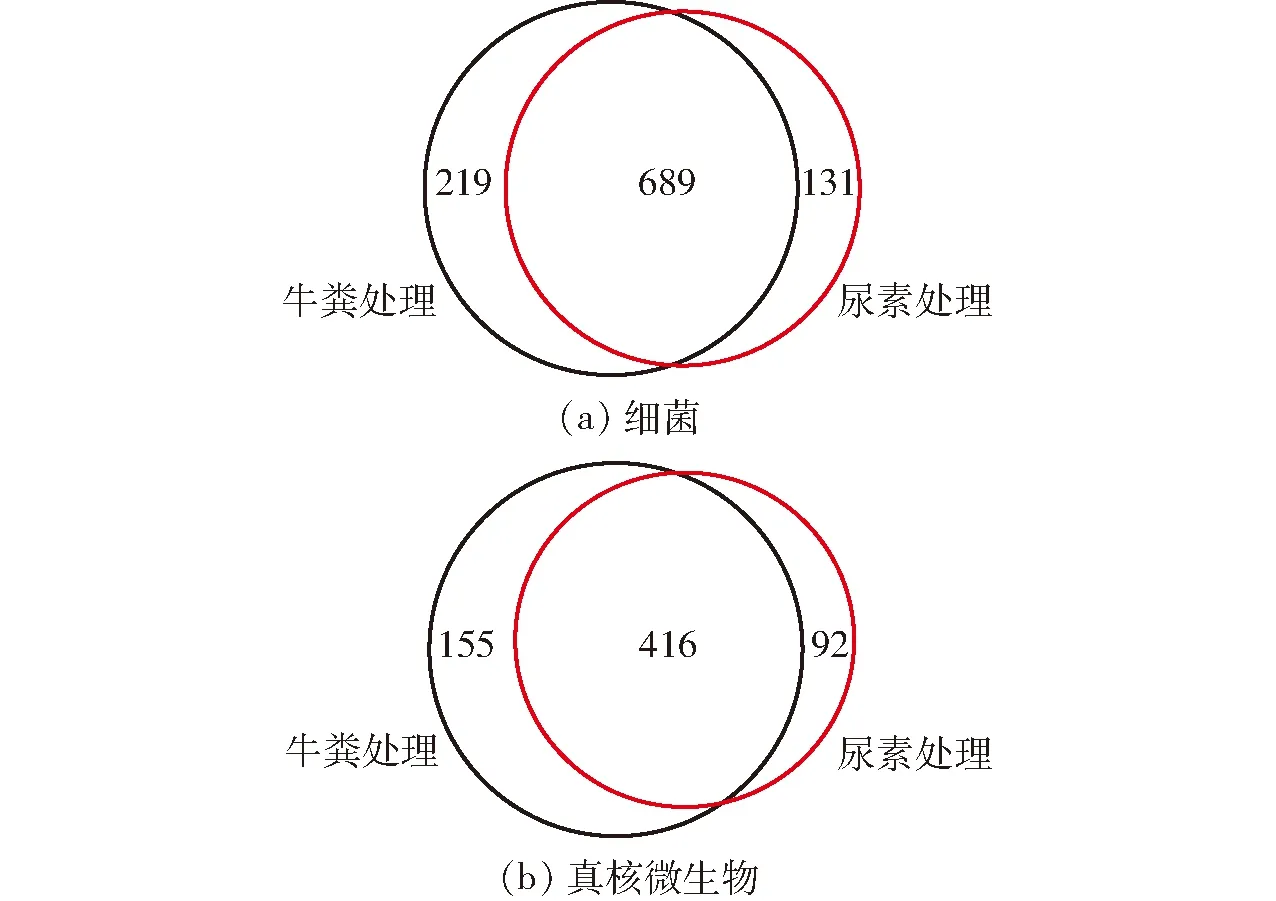
图4 两种粗制肥料中细菌和真核微生物群落OTU的维恩图Fig.4 Venn diagrams of bacterial and eukaryotic microbial OTUs among two treatments
3 讨论
本文较已有关于水稻秸秆堆肥粗制肥料的相关研究获得了更多关于粗制肥料中微生物群落结构的信息。RATRI等[20]应用变性梯度凝胶电泳(DGGE)检测方法,研究了水稻秸秆堆肥处理中微生物群落的结构,并确定了12个属的细菌优势类群。FUENTES等[21]应用焦磷酸测序方法,研究了堆肥不同时期的真菌群落结构,并获得了20个属的真菌类群信息。而本文应用基于Illmina平台的高通量测序方法,通过微生物物种注释共获得细菌292个属、真菌67个属、原生动物55个属和藻类35个属,为水稻秸秆堆肥制得的粗制肥料中微生物群落结构的解析提供了更加全面的数据。
添加尿素的水稻秸秆经堆肥发酵后,其粗制肥料中细菌群落分布在19个门、49个纲、99个目、179个科、257个属。添加牛粪制得的粗制肥料中,细菌群落分布在22个门、53个纲、104个目、187个科、276个属。然而,两种粗制肥料中细菌群落中的优势菌群构成并不同,具有明显的差异。在细菌的属分类水平上,所占比例大于1%的类群中,添加尿素制得的粗制肥料中有6个细菌属,占总细菌群落75%,包括Streptomyces、Cellulosimicrobium、Pediococcus、Paenibacillus、Lactobacillus和Nonomuraea。而添加牛粪制得的粗制肥料中也有6个细菌属所占比例大于1%,占总细菌群落的60%,包括Pseudomonas、Carnobacterium、Mobilitalea、Marinospirillum、Proteiniphilum和Advenella。由此可见,两种粗制肥料中并不存在共有的、所占比例大于1%的细菌类群。该结果说明水稻秸秆堆肥发酵过程中,无机氮素和有机氮素的添加虽然可以使水稻秸秆堆肥达到相似的腐熟效果,但是不同的氮源彻底地改变了水稻秸秆堆肥制得肥料中细菌群落的构成。尿素可以促使细菌群落放线菌门中的Streptomyces和Cellulosimicrobium两个属绝对优势的生长,占总细菌群落的62%。而牛粪可以促使细菌群落变形菌门中的Pseudomonas属绝对优势的生长,占总细菌群落的47%。
已有研究指出,在秸秆堆肥腐熟初期细菌放线菌门已经存在,但它们的数量和种类相对较少。而随着堆肥腐熟的进行,特别进入高温期后堆肥腐熟料中的放线菌门细菌种类和数量均明显增多,是主要的纤维素和木质素降解者[22-23]。本文中,添加尿素样品中所占比例大于1%的6个细菌属中,有3个细菌属属于放线菌门(Streptomyces、Cellulosimicrobium、Nonomuraea),且占总细菌群落的64%。这说明尿素添加的秸秆堆肥在高温发酵期可能促进细菌放线菌门的发酵,从而更好地降解木质素,因此添加尿素的秸秆腐熟后木质素含量较低。而假单胞菌Pseudomonas是经常被分离到的具有高效纤维素分解能力的细菌,但是其最适生长温度低于40℃[24]。因此,堆肥物料在腐熟的初期和后期,经常可以分离该细菌属菌株。本文中,添加牛粪样品中假单胞菌Pseudomonas的比例高达47.8%,这很可能表明添加牛粪处理在腐熟的初期和后期是其进行纤维素分解的主要时期。
添加尿素的水稻秸秆经堆肥发酵后,其粗制肥料中真核微生物类群分布在28个门、61个纲、99个目、105个科、143个属。添加牛粪的样品中真核微生物类群分布在26个门、66纲、104目、115科、152个属。两种粗制肥料真核微生物中的优势类群并不同。在真核微生物的属分类水平上,所占比例大于1%的类群中,添加尿素制得的粗制肥料样品中有7个属,占总的真核微生物类群的61%,包括Pichia、Scopulariopsis、Mortierella、Dunaliella、Aspergillus、Conidiobolus和Thermomyces。而添加牛粪制得的粗制肥料中也有7个属所占比例大于1%,占真核微生物类群的55%,包括Aspergillus、Mucor、Thermomyces、Pichia、Absidia、Dunaliella和Conidiobolus。在大于1%比例上,两种粗制肥料样品中共有的真核微生物类群包括Pichia、Dunaliella、Aspergillus、Conidiobolus和Thermomyces5个属,占真核微生物类群总数的47.4%。该结果说明水稻秸秆发酵过程中,无机氮素和有机氮素的添加不会改变粗制肥料中优势真核微生物的群落结构,因此两种不同的堆肥处理中真核微生物可能发挥着相同且固定的作用。
RATRI等[20]研究了水稻稻秆堆肥过程中不同阶段的真菌类群,结果表明,堆肥初期、高温期以及腐熟期均以真菌子囊菌门Ascomycota为主,而高温期发现了担子菌门Basidiomycota、接合菌门Zygomycota和藻类。本研究中添加尿素和牛粪两种氮素制得的粗制肥料中真核微生物类群以真菌子囊菌门Ascomycota 为主,分别占56.3%和40.2%;其次是接合菌门Zygomycota,分别占3.8%和20.3%;藻类在两处理中分别占5.4%和2.3%;担子菌门Basidiomycota含量较低,在两处理中分别占0.3%和1.5%。而原生动物在两种处理中仅占0.3%和0.4%。由此可见,真菌子囊菌门Ascomycota 是两种粗制肥料中真核微生物的主要类群,而牛粪处理中出现了占较大比例的接合菌门Zygomycota。同时,藻类、担子菌门(Basidiomycota)真菌和原生动物在稻麦秸秆发酵堆肥中并不占优势,且不同氮源的摄入对这些真核微生物类群的影响也并不大。
已有研究结果表明,堆肥过程中真菌群落变化随时间的变化规律明显[25]。与细菌群落相比,真菌耐受温度较低[26]。因此,真菌群落在堆肥经历了高温腐熟期后才会变得更为丰富。在堆肥发酵过程中,丝状真菌菌丝的机械穿插作用可以对物料施加一定的物理破坏作用,促进进一步的物质分解[26]。比如在添加牛粪制得的粗制肥料中较为优势的Aspergillus、Mucor、Thermomyces和Absidia属真菌,均具有发达的菌丝。同时,真菌分泌胞外酶,加快有机物水解,在物理化学作用下,难降解有机物的分解效率提高[27]。比如在添加牛粪制得的粗制肥料中较为优势的Aspergillus,具有非常强的细胞胞外酶分解能力[27-28]。其大量的存在很可能会促进添加牛粪制得的粗制肥料中水解纤维素酶、木质素酶、蛋白质酶、脂酶等胞外酶的产生,进而促进水稻秸秆的腐熟发酵。
4 结束语
添加不同的氮源均可以使水稻秸秆充分地腐熟发酵,但也导致粗制肥料中发挥作用的微生物群落构成的差异。无机氮形式的尿素可以促进秸秆堆肥中的链霉菌属Streptomyces细菌大量的繁殖,进而促进该堆肥在高温期的腐熟发酵,使纤维素和木质素充分发酵。有机氮源形式的牛粪添加促使假单胞菌属Pseudomonas细菌的大量繁殖,进而会促进该堆肥在堆置初期和发酵后期进行腐熟;同时牛粪造成的真核微生物多样性分化,使真菌子囊菌门Ascomycota 和接合菌门Zygomycota中的丝状真菌大量繁殖,其菌丝的物理作用以及各种胞外酶均会促进堆肥的发酵。
