非综合征型聋患者及家庭成员耳聋基因MYO15A检测及其产前诊断意义*
2018-07-28柴福赵海亮李胜林晋业魏建芳邱书奇
柴福 赵海亮 李胜 林晋业 魏建芳 邱书奇
在遗传性耳聋中,70%为非综合征型感音神经性聋(nonsyndromic sensorineural hearingloss,NSHL),30%为综合征型聋[1]。到目前为止,已报道的耳聋基因位点已超过100个,已鉴定出70多个非综合征型耳聋位点[2]。最常见的NSHL致病基因按出现的频率依次为GJB2、SLC26A4、MYO15A、 OTOF和CDH23[3]。国外有研究表明伊朗先天性重度聋患者中MYO15A突变率约为5.71%,在土耳其则为9.9%,这些研究表明MYO15A基因突变是导致耳聋相对重要的原因[4,5]。国内报道集中于人类常见的耳聋基因GJB2、SLC26A4和线粒体基因12SrRNA突变,而且依靠基因特异性的Sanger基因测序方法检测。MYO15A基因突变是全球最常见的NSHL致病因素之一,由于基因尺寸大,且在大多数种族人群中的突变频率和频谱未知,导致MYO15A基因的诊断用常规的方法难以实现。
芯片捕获高通量测序(targeted genomic capturing and next-generation sequencing,targeted DNA-Hiseq)可以快速、高效地分析人类疾病的致病基因突变[2],本研究拟运用芯片捕获高通量测序,对2个汉族双亲正常的NSHL聋儿及家庭成员进行127个耳聋基因编码区及邻近剪切区的DNA测序,并对其中一个家庭的高危胎儿进行了孕中期的产前诊断,以探讨NSHL患者及家庭成员耳聋基因MYO15A的突变位点及该基因检测分析用于产前诊断的可行性。
1 资料与方法
1.1研究对象 2016年11月和2016年12月就诊于深圳市龙岗区耳鼻咽喉医院耳科门诊的2个NSHL患儿及家庭成员(编号为NSHL-01和NSHL-02)。2个家庭无亲缘关系,患儿父母亲均表型正常,非近亲结婚。本研究经深圳市耳鼻咽喉研究所伦理委员会批准,所有基因诊断和产前基因诊断工作均取得患者及家属的同意并签署知情同意书。
NSHL-01: 病例1,男, 2岁8月,足月顺产,其父母听力正常且无身体其它部位异常,家族中无类似耳聋患者,无耳毒性药物使用史。耳部检查:耳廓及外耳道发育正常,鼓膜未见异常;发育及智力正常,无其它既往疾病史;40 Hz相关电位和听性脑干反应(ABR)未引出波形,颞骨CT未发现异常;临床诊断为非综合征型感音神经性聋(NSHL),并已为患儿行人工耳蜗植入术。
NSHL-02:病例2,男,3岁6个月,足月顺产,生长发育史正常。其父母听力正常且无其它异常,家族中无类似耳聋患者,无耳毒性药物使用史。出生后听力筛查未通过,确诊为双侧对称性中度感音神经性聋;10月龄开始佩戴助听器,语言发育好。耳部检查:双耳廓及耳道发育正常,鼓膜正常,发育及智力正常,身体其它部位无异常。患儿3岁时双侧行为测听0.5、1、2、4 kHz气导平均听阈为55 dB HL, 颞骨CT未发现异常。临床诊断为非综合征型感音神经性聋(NSHL);患儿母亲已再次妊娠。
1.2研究方法
1.2.1基因组DNA的提取 签署知情同意书后,抽取患儿及其家庭成员外周静脉血各2 ml,含乙二胺四乙酸(ethylene diamine tetracetic acid EDTA)的抗凝管保存备用。应用我国TIANGEN BIOTECH公司 Blood DNA提取试剂盒提取样本DNA,操作步骤按照试剂盒说明书进行。NSHL-02家庭母亲妊娠18周时,超声引导下经腹抽取胎儿羊水10 ml,进行产前基因诊断。
1.2.2目标区域捕获测序 应用已知遗传性聋基因捕获试剂盒,对遗传性聋基因编码区及临近剪切区的DNA进行捕获和富集,靶向的基因见表1;具体方法:样本准备及外显子捕获,建立测序文库:准备3-5耳聋基因组DNA,用自适应高聚焦超声技术,将基因组DNA随机打成200~300 bp的片段,打断的DNA进行修复,末端加碱基A,并接上接头;聚合酶链反应(PCR),建立测序文库;扩增后进行磁珠纯化,制备杂交文库;文库鉴定合格后的DNA片段与GenCap耳聋基因捕获试剂盒(GenCap DeafPanel)进行杂交,包含127个耳聋基因和线粒体基因,这个芯片能够捕获所有外显子和两侧的内含子(10 db)序列。将经过富集的测序区域DNA洗脱下来,PCR扩增建成测序文库。
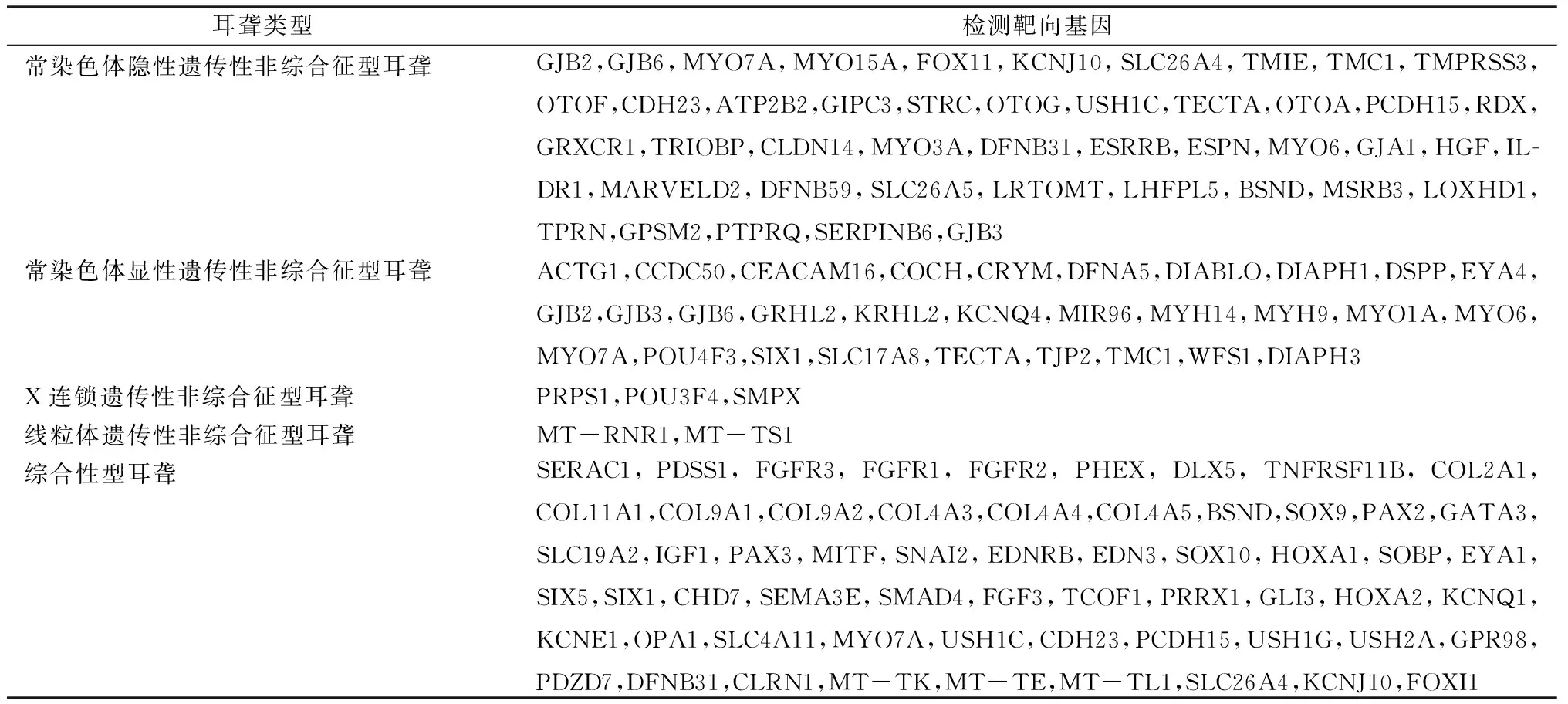
表1 不同类型耳聋检测的靶向基因
1.2.3Hiseq2000高通量测序生成成对的reads(PE90)[1]采用illlumina Hiseq2000高通量测序系统,主要通过把含有耳聋目标基因DNA的随机片段杂交到含有接头的探针上,对富集到耳聋基因的DNA片段,通过延长与桥梁扩增,用4种末端被封闭的、由不同荧光素标记的碱基进行边合成边测序,此过程覆盖了目标序列碱基>98%的区域,对碱基位置的检测准确度>99%,平均深度不小于200X(本实验在深圳华大基因完成)。
1.2.4数据分析及突变命名 对检测到的碱基变异在NCBIdbSNP数据库、Hapmap数据库和1000基因组数据库上进行分析,排除已知多态性位点。通过检索人类基因突变数据库Human Gene Mutation Datebase(HGMD,http://www.hgmd.org/)和Pubmed数据库(http:www.cnbi.nlm.nih.gov/pubmed/)来明确变异是否为已知致病突变;用Mutation Tater、SIFT和Polyphen软件对新变异进行蛋白功能预测,并对200例健康无关个体测序以排除多态性位点。新突变的命名参照国际基因变异命名体制(http://www.hgvs.org/mutnomen)提供的命名法来命名。
1.2.5桑格(sanger)测序 使用sanger测序对包含潜在突变的DNA序列进行分析来确认该变异,即使用家系成员的基因组DNA进行验证,本研究2个家庭均有父母的基因组DNA进行验证。
1.2.6胎儿产前诊断 NSHL-02家系中先证者的致病基因变异明确后,其母亲孕18周时采集胎儿羊水标本行产前诊断,提取DNA并用桑格测序法对胎儿进行该基因变异分析。用ADI基因测序仪进行DNA片段分析,需排除母体污染,排除方法:运用美国Gene Napper V3.2软件,污染标准:胎儿各等位基因位点萤光峰分别来自父母,胎儿各位点无来自母亲的第二荧光峰。
2 结果
2.1MYO15A基因(参考序列NM-016239)突变检测结果 病例1及家庭成员(NSHL-01):患儿检出MYO15A基因内含子18亚区c.5134-1G>A杂合突变和外显子20亚区c.5324A>C杂合突变,千人计划中全部测序样本中无关于此SNP的频率信息,200例正常测序样本中无关于此SNP的频率信息。 患儿的父亲(Ⅰ:1)携带c.5134-1G>A杂合突变,而患儿的母亲(Ⅰ:2)携带c.5324A>C杂合突变(图1)。
病例2及家庭成员(NSHL-02):在患儿检出MYO15A基因第二外显子c.374delG杂合突变和外显子56亚区c.9358C>T杂合突变,千人计划中全部测序样本中无关于此SNP的频率信息,200例正常测序样本中无关于此SNP的频率信息。 患儿的父亲(Ⅰ:1)携带c.9358C>T杂合突变,而患儿的母亲(Ⅰ:2)携带c.374delG杂合突变(图2)。
2.2MYO15A基因突变预测 MYO15A基因c.5134-1G>A为剪接突变,该突变位于MYO15A基因编码的肌球蛋白15的运动结构域,可能导致mRNA的剪接过程发生异常,Mutation Taster预测可能损害编码蛋白质的功能。MYO15A基因c.5324A>C为错义突变,MYO15A基因编码肌球蛋白15,其1 775位谷氨酸替换成脯氨酸,位于肌球蛋白15的运动结构域,用SIFT和Polyphen软件对其蛋白功能进行预测,结果均为有害。MYO15A基因 c.374delG为框移突变,该突变位于肌球蛋白15的N末端结构,导致其编码的肌球蛋白15序列第125精氨酸突变之后还是变成了精氨酸,使终止密码子提前到319位,产生截短蛋白或被降解。

图1 NSHL-01家庭 MYO15A基因新的突变(参考序列NM-016239) 通过病例1父母的血液DNA测序,验证突变,a:c.5134-1G>A杂合突变,是遗传其父亲的;b:c.5324A>C杂合突变,是遗传其母亲的。

图2 NSHL-02家庭 MYO15A基因新的突变(参考序列NM-016239) 通过病例2父母的血液DNA测序,验证突变,a:c.374delG杂合突变,是遗传其母亲的;b:c.9358C>T杂合突变,是遗传其父亲的。
MYO15A基因c.9358C>T,为无义突变,该突变位于肌球蛋白15的第二个肌球蛋白尾部同源物4(MyTH4)的结构域,导致其编码的肌球蛋白15序列第3 120位谷氨酰胺变为终止密码子,该突变可能导致编码蛋白序列提前终止,产生截短蛋白或提前终止。
2.3胎儿产前诊断结果 应用sanger测序法分析NSHL-02家庭中胎儿MYO15A基因突变情况,发现胎儿携带c.9358C>T杂合突变,不携带c.374delG杂合突变,表型为正常的可能性大。经遗传咨询后,母亲选择继续妊娠,胎儿足月分娩后,采取足跟血进行基因诊断,结果与产前诊断一致,且婴儿发育正常,已通过新生儿听力筛查。
3 讨论
MYO15A基因突变是全球导致非综合征型耳聋常染色体隐性遗传最常见的原因之一[2]。第一次报道的DFNH3表型听力损失病例是印度尼西亚一个偏僻村庄的患者,该村庄有2%的人口有听力损失[6];随后,研究发现该村庄两个没有血缘关系的家庭的感音性听力损失者主要基因突变与肌球蛋白15A(MYO15A)基因有关[7]。已报道有192个MYO15A的隐性突变与听力损失有关[8],本研究中确定的四个突变是新发现的,分别是病例1 MYO15A基因的c.5134-1G>A和c.5324A>C以及病例2 MYO15A基因的c.9358C>T和c.9358C>T,国内外尚未报道。
MYO15A基因是在17p11.2染色体上,横跨71 kb,由66个外显子组成,编码3 530个氨基酸的蛋白质,即肌球蛋白15A。MYO15A基因突变会引起常染色体非综合征型感音神经性聋,其分子包括三个进化上相对保守的区域(头部、颈部和尾部);头部区域包括N末端结构域和运动结构域,运动结构域负责ATP活性,并且包含ATP和肌动蛋白的两个结合位点;颈部区域包含钙调蛋白轻链结合的IQ基序;尾部区域包括2个MyTH4(肌球蛋白尾部同源物4)结构域,2个FERM结构域,1个SH3(src同源3)结构域,C末端亚型I和PDZ的结合基序[7]。MYO15A在内耳毛细胞立体细胞的分化和伸长中的作用非常重要,对于毛细胞中的肌动蛋白组织形成也是必需的[9]。
纯合子shaker-2转基因小鼠中发现,在小鼠发育早期,由于该结构域中的终止密码子的提前出现,从而导致在shaker-2转基因小鼠中MYO15A的缺失,这些小鼠表现出严重的耳聋,类似于在人类DFNH3患者中观察到的耳聋表征[10,11]。本研究中,病例1的MYO15A基因复合突变(c.5134-1G>A的剪接突变和c.5324A>C错义突变)位于MYO15A的运动结构域中,该结构域包含ATP和肌动蛋白结合位点,在体外产生移动肌动蛋白丝的能量;这些突变对蛋白质的功能具有有害影响。
MYO15A基因的第二个外显子巨大,目前发生在第二外显子上的突变数也是最多的;但发生在第二外显子上的一些突变临床表现为中度听力损失[8];本研究病例2 MYO15A基因复合突变包括c.374delG的框移突变和c.9358C>T的无义突变,c.374delG的框移突变位于MYO15的N末端结构,产生截短蛋白;该患儿的言语频率平均听阈为55 dB HL。c.9358C>T的无义突变位于第二个MyTH4结构域;MyTH4结构域的氨基酸残基非常保守,该结构域的突变对MYO15A的正常结构和功能有显著影响,从而引起的DFNH3型聋较为严重[12]。
在常染色体非综合征型感音神经性聋患者家族中,经常涉及到GJB2和SLC26A4两个基因突变,而且许多研究只集中在这两个基因;在汉族人群中也有关于MYO15A基因突变的报道,均为应用芯片捕获高通量测序技术进行的检测,分别报道为p.Gly1441Val和p.Arg2923*等突变[13,14];而本研究在两个NSHL患儿及家庭成员中发现MYO15A基因4个新发现的突变;故在未来的基因序列突变分析中,需要确定MYO15A基因突变的确切突变频率,在进行听力损失患者的遗传学检测中应密切关注该基因;同时携带MOY15A基因突变的患儿父母再次怀孕时,应通过抽取绒毛膜细胞或羊水进行产前基因诊断,有助于预防缺陷儿的出生。
