柱前衍生高效液相色谱法测定血管紧张素的氨基酸组成
2018-07-26丁金国李晓倩黄臻辉
丁金国 李晓倩 黄臻辉
摘 要 目的:建立一种测定血管紧张素氨基酸组成的柱前衍生高效液相色谱法。方法:血管紧张素经酸水解产生的游离氨基酸,用AQC衍生后,以醋酸钠溶液和乙腈作为流动相,流速为1.0 ml/min,经AccQ·Tag色谱柱梯度洗脱,于波长254 nm处测定。通过外标法计算样品中氨基酸组成。结果:His、Arg、Pro、Tyr、Val、Phe在浓度范围0.02~1.00 mmol/ml内与峰面积呈良好的线性关系,Asp在0.02~0.75 mmol/ml浓度范围内与峰面积呈良好的线性关系。结论:本实验建立的分析方法分离效果较好,可用于血管紧张素氨基酸组成测定和肽含量测定。
关键词 血管紧张素 氨基酸组成 肽含量 HPLC
中图分类号:O657.72; TQ460.72 文献标志码:A 文章编号:1006-1533(2018)11-0098-05
Determination of amino acid composition of angiotensin amide by HPLC with precolumn derivation
DING Jinguo*, LI Xiaoqian, HUANG Zhenhui(SPH NO.1 Biochemical & Pharmaceutical Co., Ltd., Shanghai 200240, China)
ABSTRACT Objective: To establish an HPLC method with precolumn derivation to determine amino acid composition and peptide content of angiotensin amide. Methods: The free amino acids were derivatized with AQC after angiotensin amide was hydrolyzed by hydrochloric acid. The derivatives were eluted by gradient solutions composed of sodium acetate solution and acetonitrile on HPLC at flow rate of 1.0 ml/min and detective wavelength of 254 nm. The contents of amino acids in angiotensin amide were calculated based on external standard. Results: The standard curves were linear over the concentration range of 0.02~1.00 mmol/ml for His, Arg, Pro, Tyr, Val, and 0.02~0.75 mmol/ml for Asp. Conclusion: The method is suitable for the analysis of amino acid composition and peptide content of angiotensin amide.
KEy WORDS angiotensin amide; amino acid composition; peptide content; HPLC
氨基酸是多肽藥物最基本的组成单位,氨基酸组成分析可以测定多肽药物的氨基酸组成,确定多肽的肽含量,初步确定多肽药物的化学结构,对评价多肽药物质量具有重要意义[1]。进行多肽类药物的氨基酸组成分析,需先将样品水解成游离氨基酸,但大多数氨基酸不含有生色基团,无法用常规的紫外检测器直接检测。在此情况下,通常可采用高效阴离子交换色谱-积分脉冲安培法直接分离、测定[2],或将游离氨基酸转化为具有较强紫外或荧光吸收的衍生物,使其有利于测定或分离。AQC(6-氨基喹啉-N-羟基琥珀酰亚胺碳酸盐)能与一、二级氨基酸在柱前进行衍生化,反应迅速、产物稳定,并可通过紫外检测器检测。
血管紧张素能直接作用于动脉血管平滑肌上的特异性受体,使小动脉强力收缩,升压活性高于去甲肾上腺素[3-6];它也是一种超短效的静脉注射用血管收缩剂,起效快,作用消除也快。可快速精确地控制血压,而治疗剂量无快速耐受性,注射部位渗漏不会引起组织坏死,给药期间无心悸、心律失常、尿量减少等不良反应。因此,它在手术麻醉,特别是产科麻醉、高血压患者麻醉、心脏手术、败血性休克急救及血管紧张素转化酶抑制剂中毒治疗中有重要作用[7-11]。我们对血管紧张素进行了质量研究,建立了基于多肽工作对照品的HPLC含量测定方法,但不能测定氨基酸组分[12]。氨基酸组成分析研究是评价多肽药物结构的重要手段,血管紧张素是由7种氨基酸组成的8肽药物,鉴于其不含Cys(半胱氨酸)和Try(色氨酸),可采用酸水解法进行水解,并以AQC为柱前衍生化试剂,应用HPLC-UV法建立血管紧张素的氨基酸组成分析方法。氨基酸组成分析方法也可用于含量测定,无需多肽工作对照品,可丰富血管紧张素含量测定手段。
1 材料和方法
1.1 材料
血管紧张素(自制);混合氨基酸标准溶液(各氨基酸含量除Cys为1.25 mmol/ml外,余均为2.5 mmol/ml)、AccQ·Tag色谱流动相A、B和衍生试剂盒(包括AQC衍生试剂粉末及稀释剂、硼酸缓冲液)均购自Waters科技有限公司。
1.2 仪器
Waters 2695高效液相色谱仪、自动进样器、Waters 2489型UV/Vis检测器和Empower 2数据处理系统(Waters科技有限公司);XS-205 DU十万分之一天平(梅特勒-托利多公司);TYXH-1旋涡混合器(上海比朗仪器有限公司);CS101-2E鼓风干燥烘箱(重庆四达科学仪器有限公司);真空干燥箱(上海一恒科技有限公司)。
1.3 方法
1.3.1 色谱条件
色谱柱:AccQ·Tag(3.9 mm×150 mm, 5 mm);检测波长254 nm;柱温34 ℃;进样量10 ml。流动相A:140 mmol/L醋酸钠水溶液(磷酸和三乙胺调节pH为5.0);流动相B:乙腈。线性梯度洗(v/v)脱程序为:0 min,0% B;16 min,8% B;18 min,9% B;24 min,18% B;32 min,33% B;34 min,33% B;35 min,100% B;37 min,100% B;38 min,0% B;45 min,0% B。流速:1 ml/min。
1.3.2 样品水解
精密称取血管紧张素样品15.0 mg,置试管中,加6 mol/L盐酸溶液(含0.1%苯酚[2])6 ml,并在冰盐浴中放置,反复充氮气后抽真空将试管熔封,于110 ℃水解24 h后,取出放冷,加高纯水定容至50 ml。精密量取1 ml水解液,真空干燥并清洗除去残余的酸液后,用高纯水溶解定容至10 ml。
1.3.3 氨基酸衍生方法
1)衍生剂溶液 将烘箱预热至55 ℃。轻弹衍生试剂盒中2A瓶,确保所有的AQC衍生试剂粉末完全落在瓶底,移取2B瓶中衍生试剂稀释剂1 ml加入2A瓶中,加盖密封,振摇10 s,置烘箱中加热,直至衍生试剂粉末完全溶解,备用。取清洁衍生管1根,加硼酸盐缓冲液80 ml,再加上述制备的衍生剂溶液20 ml,涡旋混合10 s即得。
2)氨基酸标准溶液衍生 吸取100 ml混合氨基酸标准溶液,加0.9 ml高纯水稀释,混匀备用。取清洁衍生管,加入已稀释的混合氨基酸标准溶液10 ml,再加硼酸盐缓冲液70 ml,涡旋混合,在涡旋状态下加入衍生剂溶液20 ml,并保持涡旋混合10 s,在室温放置1 min,将衍生管封口置55 ℃烘箱中10 min,取出备用。
3)样品水解溶液衍生 取清洁衍生管,加入樣品水解溶液20 ml、硼酸盐缓冲液60 ml,涡旋混合,在涡旋状态下加入衍生剂溶液20 ml,并保持涡旋混合10 s,在室温放置1 min,将衍生管封口置55 ℃烘箱中10 min,取出备用。
2 结果
2.1 色谱条件优化及适用性评价
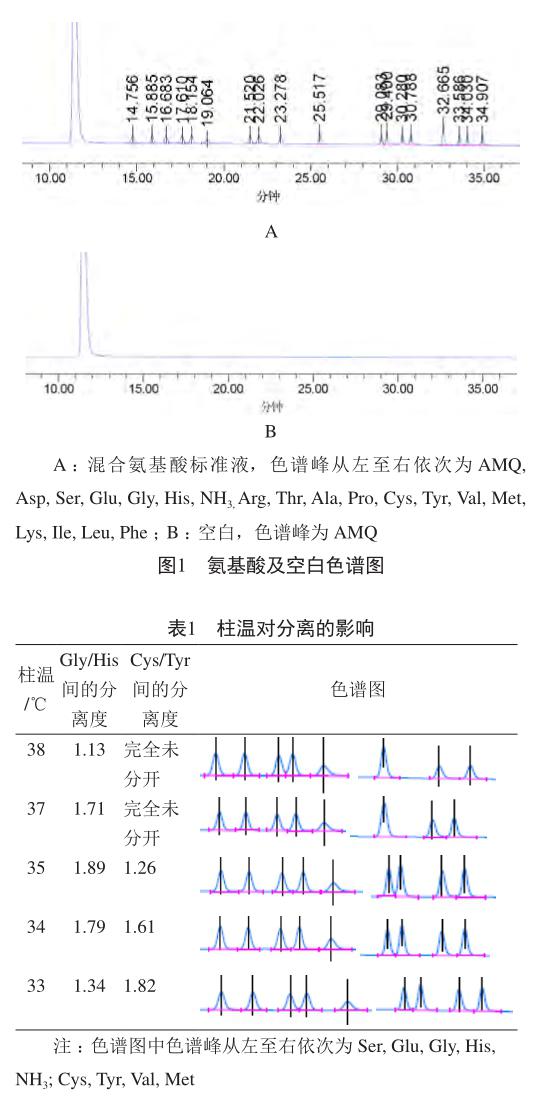
对混合氨基酸标准液和空白进行衍生处理后,分别进样,如图1所示,从从左至右依次为衍生剂水解的AMQ峰、Asp、Ser、Glu、Gly、His、NH3、Arg、Thr、Ala、Pro、Cys、Tyr、Val、Met、Lys、Ile、Leu、Phe。依1.3.1项下色谱条件,各氨基酸峰的理论塔板数均不低于20 000,分离度均大于1.5。在该色谱系统条件中除流动相配比的变化外,柱温对各氨基酸色谱峰之间的分离度有很大的影响。在研究中发现,柱温37 ℃条件下,Cys和Tyr色谱峰完全重合。通过柱温的改变发现柱温较高时,Gly/His的分离度有所增加,但Cys/Tyr间的分离度变差,甚至完全不能分离,温度降低,Gly/His的分离度降低,但Cys/Tyr的分离度增加,通过不断的摸索发现在34 ℃条件下,各氨基酸色谱峰的分离度均能达到要求(表1)。
2.2 线性关系、重复性与样品溶液稳定性试验
配制浓度为0.02、0.25、0.50、0.75、1.00 mmol/ml的系列混合氨基酸标准溶液。对各浓度溶液进行衍生化处理后,进样测定峰面积。以氨基酸浓度对峰面积绘制标准曲线(表2)。结果表明,除天冬氨酸(Asp)之外,其余6种氨基酸在0.02~1.00 mmol/ml浓度范围内与峰面积呈良好的线性关系;天冬氨酸在浓度较高的时候线性关系较差,而在0.02~0.75 mmol/ml浓度范围内与峰面积呈良好的线性关系。
取供试品溶液,衍生,重复进样6针,测定峰面积,以Asp、His、Arg、Pro、Tyr、Val、Phe氨基酸的峰面积计算RSD,分别为1.09%、1.11%、1.17%、1.07%、1.10%、1.10%、1.07%。结果表明本法对同一样品重复测定的精密度良好。将衍生后的同一混合氨基酸标准溶液分别于0、3、6、9、12、15 h进样,测定峰面积,计算得各氨基酸的RSD分别为1.09%、1.11%、1.17%、1.07%、1.10%、1.10%、1.07%。结果显示衍生后的混合氨基酸标准溶液在15 h内基本稳定。
2.3 样品水解条件优化与氨基酸组成测定
多肽水解的目的是破坏肽键,产生游离的氨基酸。在试验过程中考察16 h和24 h时各氨基酸的水解情况(表3)。结果表明,在16 h时,His和Val水解程度较低,达不到理论值。因此仍采用24 h。分别取各批次血管紧张素样品衍生溶液10 ml注入液相色谱仪,记录色谱图(图2),按外标法以峰面积计算各氨基酸的摩尔数。血管紧张素以Asp、Arg、His、Pro和Phe的摩尔数之和除以5作为1,计算各氨基酸的相对比值(表4)。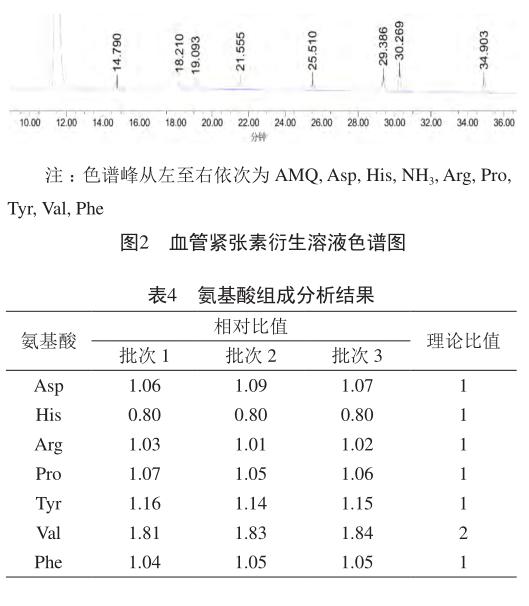
2.4 样品肽含量测定
根据上述氨基酸比值的数据以及美国药典氨基酸组成分析方法中所述,其中Asp、Arg、Phe这三个氨基酸水解情况更好,以这三个氨基酸摩尔数之和的平均数,计算肽含量。按美国药典氨基酸组成分析方法[13]中的公式100 × m/ms(其中m为测得值,ms为称量值),结果如表5所示。

3 讨论
氨基酸组成分析中干扰性因素较多,在试验之前应充分清洗钝化液相色谱,尽量减少液相系统对测定的干扰。取下色谱柱,将各管路中的有机相用高纯水进行过渡冲洗,用30%磷酸清洗1 h左右,再换高纯水冲洗至出水管路口pH为6~7。将清洗好的管路进行钝化,用6 mol/L硝酸溶液冲洗1 h左右,再换高纯水冲洗至出水管路口pH为6~7,完成钝化。除色谱仪器的准备之外,试验中所用玻璃器具如样品水解管、衍生管等,应在试验前用1 mol/L盐酸溶液煮1 h或在浓硝酸中浸泡后,用高纯水冲洗数次,烘干放置于密闭器具中备用。试验过程应尽量无尘,并戴无尘手套和口罩,防止杂蛋白的影响。
氨基酸分析中常见的柱前衍生试剂主要有OPA(邻苯二甲醛)、FMOC-Cl(氯甲酸芴甲酯)、FITC(异硫氰酸荧光素)、AQC(6-氨基喹啉-N-羟基琥珀酰亚胺碳酸盐),其中OPA不能与仲氨酸反应;FMOC-Cl衍生产物干扰测定,需用戊烷将反应过程中剩余的衍生试剂萃取出,以终止衍生反应并避免衍生物的水解;FITC要求高温和较长时间的反应;因此选用AQC,其能与一、二级氨基酸进行快速衍生化反应,且衍生产物稳定,可通过UV检测,剩余试剂在衍生化反应过程中水解,不干扰测定[1]。研究结果表明,采用AQC作衍生化试剂的柱前衍生化高效液相色谱法具有分离度良好、重复性好等优点,能较好地测定多肽药物中氨基酸的相对比值,对多肽药物的质量控制和结构确定都有一定的意义。
在氨基酸组成测定结果已知时,如已知多肽的分子量,可通过氨基酸分析的方法进行肽含量标定。通常在氨基酸水解过程中,Trp、Cys、Thr、Ser、Met容易完全或部分破坏,Ile、Val不容易完全裂解,而有些氨基酸如Gly、Ser容易被污染而使结果发生偏差。其中能较好、稳定水解的氨基酸主要有Asp、Glu、Ala、Leu、Phe、Lys、Arg,可以用这些稳定水解的氨基酸的摩尔数计算肽含量。在研究中发现,通过液相法[12]测定血管紧张素的肽含量与氨基酸组成测定的结果稍有不同,其中通过氨基酸组成测定得到的肽含量相对较低,分析原因认为,样品在酸水解过程,以及清洗去除酸液和残渣的过程中,发生了一定程度的损失,而混合氨基酸标准液因本身由游离氨基酸组成,未经过酸水解和后处理清洗过程,故使样品测得的值较低。在之后的研究中,预采取将混合氨基酸标准液经过酸水解过程,来校正样品的损失,或通过已知肽含量的标准肽,经酸水解处理做回收率试验,以校正样品的损失。
在多肽药物质量研究的過程中,液相法测定药物含量需要对照品,有些药物含量测定用对照品不能获得,肽含量的确定存在困难。而氨基酸组成分析的方法,能很好地用于多肽药物肽含量的测定,因此在多肽药物的质量控制中有十分重要的作用。
致谢:感谢霍建丽、耿敏对本文的贡献。
参考文献
[1] Kaspar H, Dettmer K, Gronwald W, et al. Advances in amino acid analysis[J]. Anal Bioanal Chem, 2009, 393(2): 445-452.
[2] Zhou YH, Xin N, Hu YZ. HPLC with precolumn derivatization determination of amino acid composition of oxytocin[J]. Chin J Pharm Anal, 2011, 31(8): 1541-1544.
[3] Hidaka T, Tsuneyoshi I, Boyle WA, et al. Marked synergism between vasopressin and angiotensin II in a human isolated artery[J]. Crit Care Med, 2005, 33(11): 2613-2620.
[4] Paiva TB, Paiva AC. Some pharmacological actions of synthetic analogues of angiotensin amide[J]. Br J Pharmacol Chemother, 1960, 15(4): 557-560.
[5] Wray GM, Coakley JH. Severe septic shock unresponsive to noradrenaline[J]. Lancet, 1995, 346(8990): 1604.
[6] Yunge M, Petros A. Angiotensin for septic shock unresponsive to noradrenaline[J]. Arch Dis Child, 2000, 82(5): 388-389.
[7] McKinnon RP, Sinclair CJ. Angiotensinamide in the treatment of probable anaphylaxis to succinylated gelatin (Gelofusine)[J]. Anaesthesia, 1994, 49(4): 309-311.
[8] Vincent RD Jr, Werhan CF, Norman PF, et al. Prophylactic angiotensin II infusion during spinal anesthesia for elective cesarean delivery[J]. Anesthesiology, 1998, 88(6): 1475-1479.
[9] Eyraud D, Mouren S, Teugels K, et al. Treating anesthesiainduced hypotension by angiotensin II in patients chronically treated with angiotensin-converting enzyme inhibitors[J]. Anesth Analg, 1998, 86(2): 259-263.
[10] Newby DE, Lee MR, Gray AJ, et al. Enalapril overdose and the corrective effect of intravenous angiotensin II[J]. Br J Clin Pharmacol, 1995, 40(1): 103-104.
[11] Thaker U, Geary V, Chalmers P, et al. Low systemic vascular resistance during cardiac surgery: case reports, brief review, and management with angiotensin II[J]. J Cardiothorac Anesth, 1990, 4(3): 360-363.
[12] 霍建丽, 耿敏, 丁金国, 等. RP-HPLC法测定血管紧张素的肽含量及有关物质[J]. 药物生物技术, 2012, 19(6): 521-524.
[13] United States Pharmacopeial Convention. U. S. Pharmacopeia USP40 [M]. Rockville: United States Pharmacopeial Convention, Inc. 2017: 1093-1105.
