甘草酸提取废液α-葡萄糖苷酶抑制剂的筛选与鉴定
2018-07-23樊金玲程源斌朱文学
余 颖,樊金玲,,程源斌,朱文学
(1.河南科技大学食品与生物工程学院,河南 洛阳 471023;2.洛阳蓝斯利科技有限公司,河南 洛阳 471023)
α-葡萄糖苷酶(EC3.2.1.20)是人体消化碳水化合物的关键酶[1-2],催化水解低聚糖底物非还原末端的α-1,4-糖苷键,释放出α-D-葡萄糖[3-4]。α-葡萄糖苷酶抑制剂可通过抑制人体肠道内α-葡萄糖苷酶活性,延缓食物中碳水化合物的水解,从而减慢糖的吸收,降低餐后血糖水平[5-6],可用于防治II型糖尿病及其并发症[7]。由于药物合成的α-葡萄糖苷酶抑制剂的不良反应屡有报道[8],因此从天然动植物及食品中开发α-葡萄糖苷酶抑制剂,用于降血糖功能性食品及功能性食品配料具有重要意义[9-12]。
甘草是我国卫生与计划生育委员会委颁布的药食同源的多年生草本植物[13],其蕴藏量丰富,在中、西方食品和药品中均有广泛应用[14-15]。其主要的利用形式之一是提取甘草酸,提酸后的甘草药渣和废液则被遗弃,造成了极大的环境污染和资源浪费[16-17]。近年来研究表明,甘草的水提取物和乙醇提取物均对α-葡萄糖苷酶具有显著的抑制活性[18-19],其中甘草黄酮、甘草酸是主要的活性物质,例如:甘草酚、甘草异黄酮乙、甘草异黄酮甲等具有较高的α-葡萄糖苷酶抑制活性[20-25]。本实验从甘草酸的工业生产出发,对甘草酸提取废液进行梯度萃取,研究不同溶剂萃取物的α-葡萄糖苷酶抑制活性,探讨抑制活性最强的萃取相对α-葡萄糖苷酶的抑制作用类型及其与阿卡波糖的协同作用,并采用超高效液相色谱-二极管阵列检测-质谱(ultra high performance liquid chromatography with diode array detector mass spectrometry,UHPLC-DAD-MS)等多种方法对其主要成分的化学结构进行分析鉴定,同时测定了主要成分的含量。本研究为甘草及甘草酸提取废液的综合开发和利用提供理论依据。
1 材料与方法
1.1 材料与试剂
甘草酸提取废料由洛阳蓝斯利科技有限公司提供。
α-葡萄糖苷酶、阿卡波糖、甘草素、异甘草素、柚皮素、芒柄花素 上海源叶生物科技有限公司;葡萄糖测定试剂盒 中生北控生物科技股份有限公司;萃取用有机溶剂均为国产分析纯。
1.2 仪器与设备
Model 680全自动酶标仪 美国Bio-Rad公司;1260高效液相色谱仪、6520 四极杆-飞行时间-质谱(quadrupole-time of flight-mass spectrometry,Q-TOFMS)仪 美国Agilent 公司;RE52CS-1旋转蒸发仪上海亚荣生化仪器厂;SCIENTZ-10N冷冻干燥机 宁波新芝生物科技有限公司;101-2A电热鼓风干燥箱 天津泰斯特仪器有限公司。
1.3 方法
1.3.1 样品的制备
甘草酸提取废液浓缩物依次用正己烷、氯仿、乙酸乙酯和正丁醇萃取3 次(体积比1∶1),分别合并各萃取液。各萃取液和萃余水相经真空减压浓缩和真空冷冻干燥分别得正己烷、氯仿、乙酸乙酯、正丁醇萃取相和水相。冷藏备用,使用前用甲醇复溶。
1.3.2 样品对α-葡萄糖苷酶的抑制作用
测定甘草酸提取废液及其各萃取相和萃余水相对α-葡萄糖苷酶的抑制活性,其具体方法参考文献[26],并作部分修改。取α-葡萄糖苷酶溶液(50 U/mL,pH 6.86磷酸盐缓冲液溶解)40 μL于96 孔酶标板中,加入20 μL不同质量浓度(0.00、0.05、0.10、0.20、0.50、1.00、2.0 g/L)样品(甲醇溶解),置于37 ℃恒温箱中预热15 min;加入10 mmol/L麦芽糖(pH 6.86磷酸盐缓冲液溶解)40 μL,37 ℃保温反应30 min;然后加入0.1 mol/L Na2CO3100 μL终止反应。取上述反应液50 μL,加入葡萄糖测定试剂盒中的葡萄糖工作液150 μL,充分混匀,37 ℃反应30 min,于505 nm波长处检测吸光度A。每个样品重复测定4 孔,取平均值。按式(1)计算样品对α-葡萄糖苷酶的抑制率,并通过方程拟合计算半抑制浓度(half maximal inhibitory concentration,IC50)。式(1)中酶空白组、样品组、样品背景组的反应体系见表1。同时,按上述方法测定不同质量浓度阿卡波糖对α-葡萄糖苷酶的抑制率和IC50。
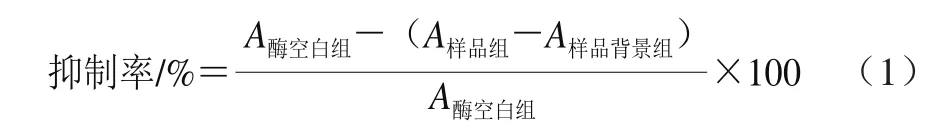

表1 α-葡萄糖苷酶活性反应体系Table1 Reaction system for α-glucosidase activity
1.3.3 样品对α-葡萄糖苷酶的抑制作用类型
分析1.3.2节实验中IC50最小的样品对α-葡萄糖苷酶的抑制作用类型。按照1.3.2节方法,设定样品质量浓度范围为0~1 g/L;在样品某一确定质量浓度下,改变反应体系中麦芽糖浓度(0~500 mmol/L),测定反应液的吸光度。根据Lineweaver-Burk双倒数曲线图,判断样品对α-葡萄糖苷酶的抑制作用类型。具体方法详见参考文献[27]。其中,Lineweaver-Burk双倒数曲线图是由酶促反应速率的倒数(1/V)对麦芽糖底物浓度的倒数(1/S)作图,x和y轴上的截距分别代表米氏常数(Km)和最大反应速率(Vmax)的倒数。
1.3.4 样品与阿卡波糖对α-葡萄糖苷酶的协同抑制作用分析1.3.2节实验中IC50最小的样品与阿卡波糖对α-葡萄糖苷酶的协同抑制作用。基于1.3.2节实验得到的样品与阿卡波糖的IC50,分别配制7 个质量浓度(0.25 IC50、0.50 IC50、1.00 IC50、1.50 IC50、2.00 IC50、2.50 IC50、5.00 IC50)的样品和阿卡波糖溶液,并按体积比1∶1混合,按1.3.2节方法测定混合液对α-葡萄糖苷酶的抑制率。按式(2)计算联合作用指数(combination index,CI);CI<1、CI=1、CI>1分别代表协同作用、加和作用和拮抗作用[28-31]。

式中:ρ1表示某一抑制率下混合液中提取物的质量浓度/(g/L);ρ2表示某一抑制率下混合液中阿卡波糖的质量浓度/(g/L);ρ1′表示相同抑制率下提取物单独作用的质量浓度/(g/L);ρ2′表示相同抑制率下阿卡波糖单独作用的质量浓度/(g/L)。
1.3.5 样品酸水解
称取1.3.2节实验中IC50最小的样品25 mg,溶于3 mL质量分数1% HCl-CH3OH溶液,再加入2 mL 2 mol/L HCl溶液,混合均匀,密封置于100 ℃沸水浴中反应130 min。取出室温冷却,用质量分数1% HCl-CH3OH溶液补充体积至5 mL,0.45 μm微孔滤膜(有机系)过滤后冷藏备用。
1.3.6 UHPLC-DAD-MS分析
对1.3.2节实验中IC50最小的样品进行UHPLC-DAD-MS分析。
色谱条件:SB-C18柱(100 mm×4.6 mm,1.8 μm);以乙腈(A)和0.1%甲酸水溶液(B)为流动相,梯度洗脱(0~20 min,5%~100% A);流速:0.4 mL/min;进样量:10 μL;柱温 25℃;DAD检测器,检测波长:λ1=276 nm、λ2=372 nm。
TOF-MS条件:电喷雾离子源;TOF-MS检测器;气体温度:350℃;干燥气体流速:5 mL/min;毛细管电压:4 000 V;碎裂电压:150 V。
1.3.7 样品主要成分的含量测定
HPLC分析:采用HPLC法测定酸水解样品中各成分的含量。色谱柱:ZORBAX SB-C18柱(250 mm×4.6 mm,5 μm);以甲醇(A)和1%冰醋酸水溶液(B)为流动相,梯度洗脱(0~20 min,25%~40% A;20~60 min,40%~100% A);流速:1.0 mL/min;进样量:10 μL;柱温30 ℃;DAD检测器,检测波长:λ1=276 nm、λ2=372 nm。
标准曲线的制作:称取一定量的标准品,用甲醇配制系列质量浓度的标准品使用液,按上述方法进行分析检测。以溶液质量浓度(甘草素质量浓度单位mg/mL,其余为μg/mL)为横坐标,以峰面积为纵坐标,进行线性回归,得到回归方程分别为:甘草素(276 nm):y=37 293x+1 381.4(R2=0.999 8);柚皮素(276 nm):y=18.193x+6.12(R2=0.999 9);异甘草素(372 nm):y=44.153x+75.745(R2=0.999 9);芒柄花素(276 nm):y=29.107x+28.13(R2=0.999 9)。
含量计算:酸水解样品中4 种主要的黄酮苷元(即甘草素、柚皮素、异甘草素以及芒柄花素)的含量按式(3)计算。
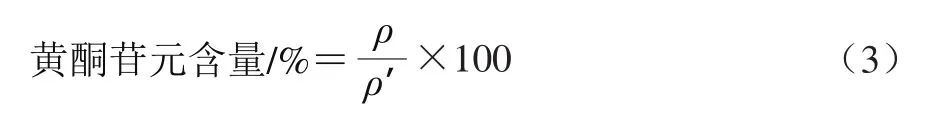
式中:ρ表示酸水解样品中黄酮苷元(甘草素、柚皮素、异甘草素、芒柄花素)的质量浓度/(mg/mL);ρ′表示酸水解样品的质量浓度/(mg/mL)。
未酸水解样品的主要化学成分为上述苷元的单糖苷和二糖苷。各黄酮苷元(甘草素、柚皮素、异甘草素、芒柄花素)的相对分子质量Mr分别以256.25、272.25、256.25、268.26计,糖苷平均分子相对分子质量Mr以240计,折合成糖苷的平均相对分子质量Mr′分别以496.25、515.25、496.25、508.26计。按式(4)计算以测定所得各苷元的含量折合成各类糖苷的总含量。
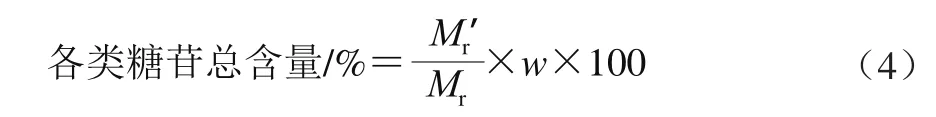
式中:Mr表示各黄酮苷元的相对分子质量;Mr′表示各黄酮苷元折合成糖苷的平均相对分子质量;w表示黄酮苷元含量/%。
2 结果与分析
2.1 甘草酸废液提取物对α-葡萄糖苷酶的抑制作用比较
甘草酸提取废液及其各萃取相和萃余水相对α-葡萄糖苷酶活性的抑制作用如图l所示。根据图1数据拟合方程得到的IC50见表2。除正己烷相外,甘草酸提取废液的各萃取相与甘草酸提取废液相比,α-葡萄糖苷酶抑制活性均有不同程度的提高。其中,抑制活性最强的是乙酸乙酯萃取相,IC50为0.152 9 g/L(得率为5.65%);其次为水相,IC50为0.618 9 g/L(得率为83.54%)。由于乙酸乙酯萃取相对α-葡萄糖苷酶的抑制活性最强,因此,后续实验均以此相为研究对象。
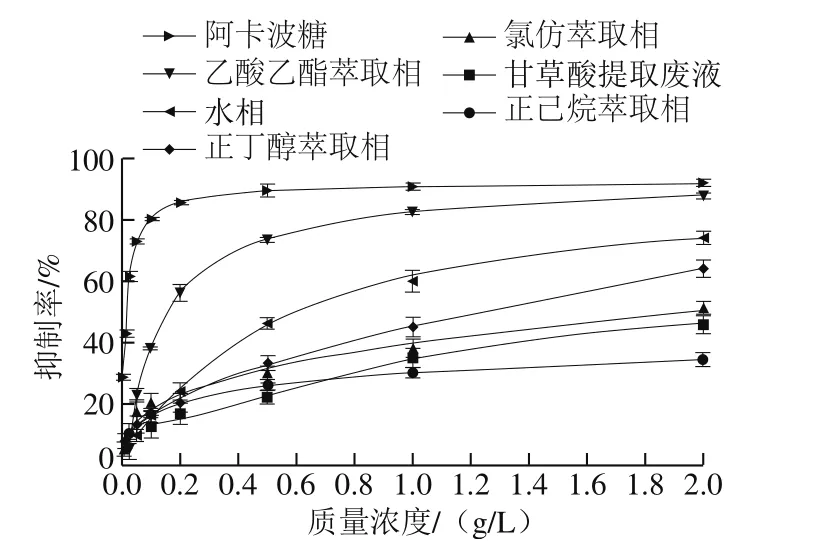
图1 甘草酸提取废液及其萃取物对α-葡萄糖苷酶的抑制作用Fig. 1 Inhibitory effect of glycyrrhizic acid extraction waste liquid against α-glucosidase
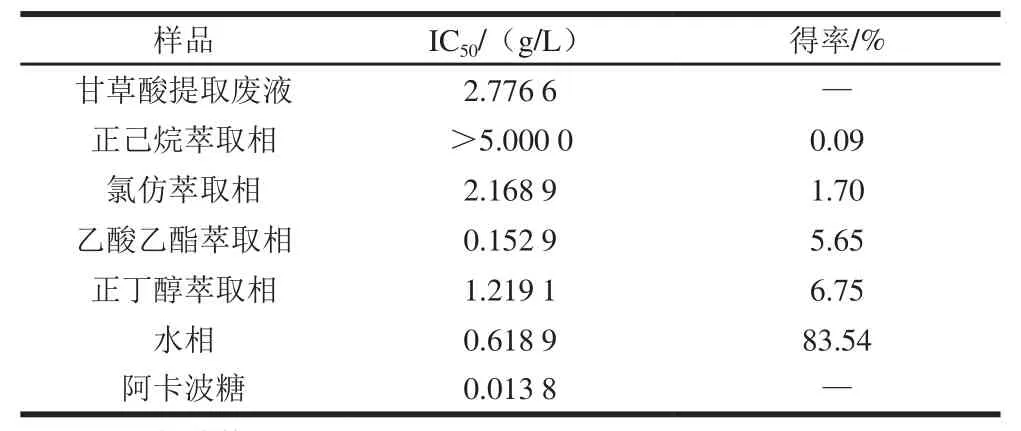
表2 甘草酸提取废液及其萃取物对α-葡萄糖苷酶的IC50和得率Table2 IC50 and yield of extracts with α-glucosidase inhibitory activity from glycyrrhizic acid extraction waste liquid
2.2 乙酸乙酯相对α-葡萄糖苷酶的抑制作用类型
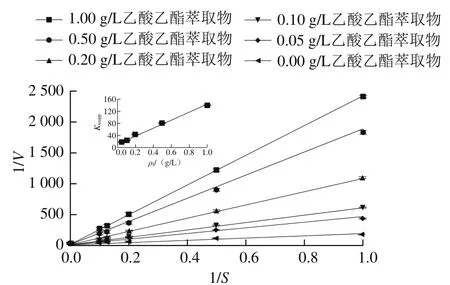
图2 乙酸乙酯提取物对α-葡萄糖苷酶抑制作用的Lineweaver-Burk双倒数图Fig. 2 Lineweaver-Burk double reciprocal plot for the inhibition of α-glucosidase by the ethyl acetate extract
按1.3.3节方法测定不同质量浓度乙酸乙酯萃取条件下麦芽糖底物浓度对α-葡萄糖苷酶催化速率的影响,根据Lineweaver-Burk双倒数作图。从图2可以看出,随着乙酸乙酯萃取相质量浓度的增大,纵轴截距不变,斜率增大,即Vmax不随着抑制剂质量浓度增大而变化,米氏常数Km随着抑制剂质量浓度的增大而不断增大,表明抑制剂质量浓度增大时,酶对底物的亲和力在不断下降,抑制剂的存在阻碍了底物和酶的结合,增加底物浓度或抑制剂质量浓度均可排除对方对酶的效应,抑制剂对酶的抑制作用表现为竞争性抑制。竞争性抑制剂只能与游离酶结合,不能与酶-底物配合物结合。以不同质量浓度抑制剂测定的表观米氏常数值(Kmapp)对抑制剂质量浓度(ρI)作图为一条直线(图2内插图),直线所得横轴截距为-Ki,即可以求得抑制常数Ki为0.085 1 g/L。
2.3 乙酸乙酯萃取相与阿卡波糖对α-葡萄糖苷酶的协同抑制作用
不同质量浓度的乙酸乙酯萃取物和阿卡波糖混合对α-葡萄糖苷酶的协同抑制作用见图3,在所测的质量浓度范围内,乙酸乙酯萃取物与阿卡波糖混合物的CI始终小于1,说明二者对α-葡萄糖苷酶存在协同抑制作用;CI呈现先增后降的趋势,表明低剂量和高剂量条件下的乙酸乙酯萃取物和阿卡波糖的协同作用大于中等剂量。
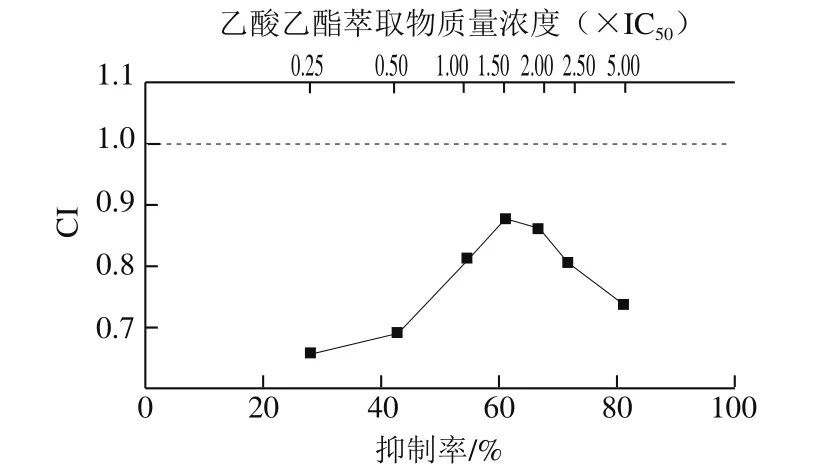
图3 乙酸乙酯萃取物和阿卡波糖混合液的CI曲线Fig. 3 CI plot of a mixture of ethyl acetate extract and acarbose
2.4 乙酸乙酯萃取相化学成分的初步鉴定

图4 乙酸乙酯提取物的UHPLC色谱图Fig. 4 UHPLC chromatograms of the ethyl acetate extract
采用UHPLC-DAD-MS对乙酸乙酯萃取相进行分析,其色谱图如图4所示。依据各色谱峰的光谱和质谱特征可看出:乙酸乙酯萃取相中的主要成分为黄酮类化合物。对其中相对丰度较高的15 个黄酮类化合物的光谱信息、质谱信息(准分子离子、特征裂片、中性丢失)进行解析,将其分为Ⅰ~Ⅴ类,初步确定了各黄酮化合物的苷元和糖基组成,详见表3,苷元结构如图5所示。

表3 乙酸乙酯萃取相的光谱、质谱特征及结构鉴定Table3 Spectral characteristics and structural identification of flavonoid compounds in the ethyl acetate extract
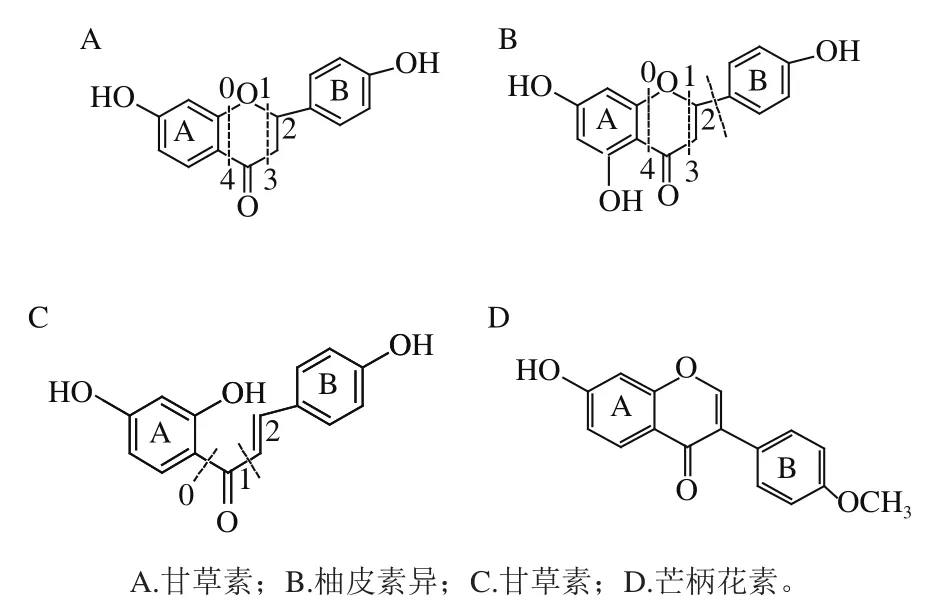
图5 不同黄酮苷元结构图Fig. 5 Structure of different flavonoid aglycones
类Ⅰ包括色谱峰1、3、6,其光谱的共同特征为:约在284 nm左右具有强的带Ⅱ吸收,带Ⅰ以肩峰形式出现或消失。此类化合物质谱的共同特征为:负离子检测模式下苷元离子的m/z为271,苷元共同的碎片离子分别为177、151和107。结合光谱图和以上特征碎片,推断类Ⅰ化合物的苷元为柚皮素,m/z 151为[1,3A-H]-,m/z 107为[0,4A-H]-,m/z 177为苷元失去B环所得,即[苷元-H-B]-。峰1和峰3是同分异构体,其[M-H]-的m/z为433,丢失1 个六碳糖中性碎片162 u得到苷元离子m/z 271;因此,峰1和峰3均为柚皮素-六碳糖单糖苷。目前尚无法判断峰1和3中糖苷键位置。峰6[M-H]-的m/z为565,丢失1 个五碳糖中性碎片132 u得到碎片离子m/z 433,再丢失1 个六碳糖中性碎片162 u得到苷元离子m/z 271;因此,峰6为柚皮素-六碳糖-五碳糖二糖苷。
类Ⅱ包括色谱峰2、4和5,其光谱的共同特征为:约在271~276 nm波长处具有强的带Ⅱ吸收,带Ⅰ以低强度峰的形式出现在313~316 nm波长处,表明此类化合物应为双氢黄酮或双氢黄酮醇。与类Ⅰ色谱峰相比,其带Ⅱ吸收向短波移动10 nm左右,表明可能不存在5-羟基。此类化合物质谱的共同特征为:负离子检测模式下苷元离子的m/z为255,苷元共同的碎片离子分别为135、119和91。结合光谱图和以上特征碎片推断此类化合物的苷元为甘草素,m/z 135为[1,3A-H]-,m/z 91为[0,4A-H]-,m/z 119为[1,3B-H]-。峰2[M-H]-的m/z为417,丢失1 个六碳糖中性碎片162 u得到苷元离子m/z 255;因此,峰2为甘草素-六碳糖单糖苷。峰4和峰5是同分异构体,其[M-H]-的m/z为549,丢失1 个五碳糖中性碎片132 u得到碎片离子m/z 417,再丢失1 个六碳糖中性碎片162 u得到苷元离子m/z 255;因此,峰4和峰5均为甘草素-六碳糖-五碳糖二糖苷。
类Ⅲ包括色谱峰7、9、10和12,其光谱的共同特征为:约在362~373 nm波长处具有强的带Ⅰ吸收,带Ⅱ以低强度峰形式出现在242~246 nm处,表明此类化合物应为查耳酮类。此类化合物质谱的共同特征为:负离子检测模式下苷元离子的m/z为255,苷元共同的碎片离子分别为135、119和91。此类苷元碎片与类Ⅱ完全相同,但此类化合物的光谱图显著不同于类Ⅱ化合物,为查耳酮化合物。结合光谱图和以上特征碎片推断类Ⅲ化合物的苷元为异甘草素,m/z 135为[1A-H]-、m/z 91为[0A-H]-、m/z 119为[1B-H]-。峰7和峰10是同分异构体,其[M-H]-的m/z为549,丢失1 个五碳糖中性碎片132 u得到碎片离子m/z 417,再丢失1 个六碳糖中性碎片162 u得到苷元离子m/z 255;因此,峰7和峰10均为异甘草素-六碳糖-五碳糖二糖苷。峰9、12为同分异构体,其[M-H]-的m/z为417,丢失1 个六碳糖中性碎片162 u得到苷元离子m/z 255;因此,峰9、12为异甘草素-六碳糖单糖苷。
类Ⅳ包括色谱峰8和11,其光谱的特征为:约在251 nm波长处具有强的带Ⅱ吸收,带Ⅰ以肩峰的形式出现在306 nm波长处,表明此类化合物应为异黄酮化合物;其带Ⅱ吸收出现在245~270 nm波长处,与双氢黄酮或双氢黄酮醇(带Ⅱ吸收出现在270~295 nm波长处)有明显区别。此类化合物质谱的共同特征为:负离子检测模式下苷元离子的m/z为267,苷元共同的碎片离子较少。结合此类化合物的光谱图,并查阅相关文献[32],推测此苷元为芒柄花素。其中,峰8的[M-H]-的m/z为561,丢失1 个五碳糖中性碎片132 u和1 个六碳糖中性碎片162 u得到苷元离子m/z 267;因此,峰8为芒柄花素-六碳糖-五碳糖二糖苷。峰11质谱图中m/z 475为[M+HCOO]-,丢失1 个六碳糖中性碎片162 u得到苷元离子m/z 267;因此,峰11为芒柄花素-六碳糖单糖苷。
类Ⅴ包括色谱峰13、14和15,其光谱的共同特征为:约在314~325 nm波长处具有强的带Ⅰ吸收,带Ⅱ以低强度峰或肩峰形式出现在281~283 nm波长处。此类化合物质谱的共同特征为:负离子检测模式下苷元离子的m/z为255,苷元共同的碎片离子分别为135、119和91,与类Ⅱ、Ⅲ相同;但此类化合物光谱图明显不同于类Ⅱ、Ⅲ化合物,其带Ⅰ吸收强于带Ⅱ吸收,出现在314、323 nm和325 nm波长处,带Ⅱ出现在281~283 nm波长处。峰13、14和15为同分异构体,其[M-H]-的m/z为725,丢失1 个六碳糖醛酸中性碎片176 u得到碎片离子m/z 549,再丢失1 个五碳糖中性碎片132 u得到m/z 417,再丢失1 个六碳糖中性碎片162 u苷元离子m/z 255;因此,峰13、14和15均为黄酮化合物的六碳糖醛酸-六碳糖-五碳糖三糖苷。尚不明确此类化合物的类别。
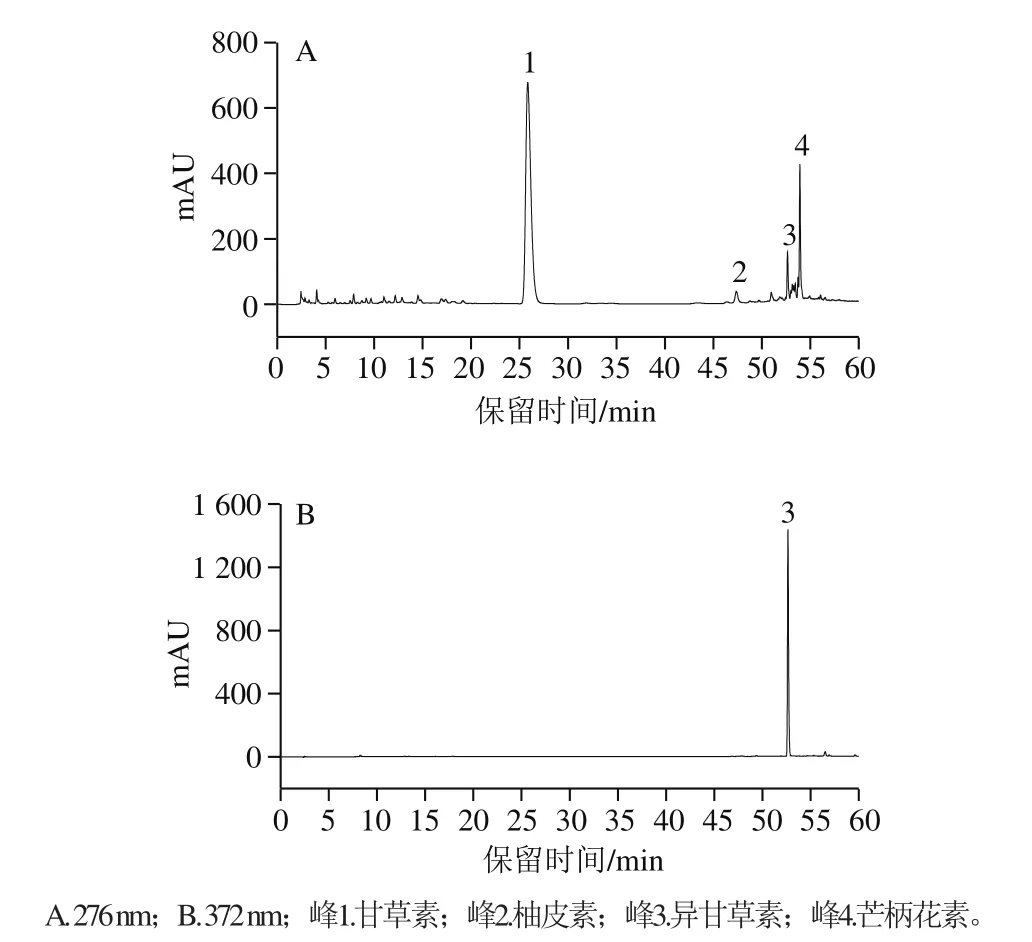
图6 乙酸乙酯提取物水解物的HPLC图Fig. 6 HPLC profile of the acid hydrolysate of the ethyl acetate extract
为了进一步确认推断的乙酸乙酯萃取相的苷元结构,将其按1.3.5节方法进行酸水解,再经HPLC分析,其色谱图如图6所示。将水解产物中主要4 个色谱峰的保留时间和光谱图与标准品进行了比较,结果表明:峰1为甘草素,峰2为柚皮素,峰3为异甘草素,峰4为芒柄花素(标准品的色谱图及光谱图略)。此结果表明苷元种类推断正确。
2.5 乙酸乙酯萃取相主要成分的含量确定
将乙酸乙酯萃取相进行酸水解,采用HPLC法测定了水解液中4 种主要的黄酮苷元(即甘草素、柚皮素、异甘草素以及芒柄花素)的含量。甘草素、柚皮素、异甘草素、芒柄花素分别为14.50%、1.56%、6.09%、2.31%。由于乙酸乙酯萃取相主要成分为上述苷元的单糖苷和二糖苷,将测定所得各苷元的含量折合成各类糖苷的总含量。甘草素类、柚皮素类、异甘草素类、芒柄花素类的黄酮含量总计分别为28.08%、2.95%、11.80%、4.38%;黄酮总含量为47.21%。
3 结 论
在甘草酸提取废液及其各萃取相和萃余水相对α-葡萄糖苷酶活性的抑制作用中,乙酸乙酯萃取相具有最强的抑制活性,IC50为0.152 9 g/L,抑制类型表现为竞争性抑制,抑制常数Ki=0.085 1 g/L,且与阿卡波糖混合对α-葡萄糖苷酶存在协同抑制作用。乙酸乙酯萃取相中的主要化学成分为黄酮类化合物,初步确定了各黄酮化合物的苷元和糖基组成为甘草素、柚皮素、异甘草素以及芒柄花素的单糖苷和二糖苷。乙酸乙酯萃取相水解液中4 种主要的黄酮苷元(即甘草素、柚皮素、异甘草素以及芒柄花素)的含量分别为:14.50%、1.56%、6.09%和2.31%。将测定所得各苷元的含量折合成各类糖苷的总含量,则甘草素类、柚皮素类、异甘草素类和芒柄花素类的黄酮含量总计分别为28.08%、2.95%、11.80%和4.38%;黄酮总含量为47.21%。
