开垦对盐渍化弃耕地土壤细菌群落组成和结构的影响
2018-07-09张玉龙姜同轩张凤华
杨 磊,张玉龙,姜同轩,张凤华
(石河子大学/新疆生产建设兵团绿洲生态农业重点实验室,新疆石河子 832003)
0 引 言
【研究意义】新疆属于干旱地区,一些不合理的灌溉制度及农业措施破坏了原有的水盐平衡,加之强烈蒸发加剧土地次生盐渍化的发生,致使大面积农田被迫弃耕。需要在弃耕地上重建次生植被来恢复土壤肥力。自2000年随着滴灌技术在新疆地区大规模应用,大面积盐渍化弃耕地得以开垦。科学合理的的开垦不仅能够提高土壤质量,还能维持绿洲生态系统的稳定。【前人研究进展】土壤微生物对于改善土壤健康具有重要的意义,是植物生长必不可少的一部分[1],其群落结构和多样性对评价土壤质量起着关键的作用[2]。在整个生态系统中土壤微生物在有机质的分解、营养物质的循环以及植物养分的有效性方面扮演着重要的作用[3]。土壤微生物群落的组成和结构受各种农业措施的影响,如耕作制度[4]、施肥管理[5]、农药使用[6]和灌溉[7]等。其中土地利用方式的变化显著改变了土壤养分以及土壤微生物群落结构和多样性[8]。在干旱区,荒地开垦为农田改变了土地利用方式和植被覆盖,引起了植被、凋落物、水分以及养分的变化。植被通过改变土壤碳氮、水分、温度等影响微生物。植被为土壤微生物提供营养物质和能量,对土壤微生物所栖息的环境产生影响。植被的存在可以增加土壤微生物量,引起土壤微生物群落多样性和结构的改变,最终影响土壤质量和功能[9]。研究发现,表层(0~20 cm)土壤中微生物种群的丰富度较高。微生物量随土层深度的增加以指数方式减少[10]。研究也表明微生物多样性随土层深度的增加而降低[11]。在新疆膜下滴灌被广泛的用于作物的生产中,在不同的生育期通过滴灌进行根外追肥,这一系列的措施可以显著地影响土壤微生物群落的分布。随着高通量测序技术的发展,像454焦磷酸测序和Illumina测序,可以为发现微生物类群提供更为直接的方法,尤其是低丰度物种的变化[12]。采用成熟的TruSeq边合成边测序技术,可以用于研究和描述土壤微生物中未培养的细菌,得到更多而又完整的微生物群落信息。运用MiSeq测序得到的生物学信息,依靠分类学原理可以将微生物类群进行更为精确的划分[13]。【本研究切入点】新疆北部大面积的农田由于不合理的管理方式导致了土地的盐碱化甚至是弃耕,限制了农业的发展。膜下滴灌技术的发展使得弃耕的土地得以开垦成农田,农田进行耕作种植不仅可以改善土壤微环境,提高土壤质量状况。与弃耕地相比,开垦后种植棉花可以显著地改变土壤细菌群落组成和结构。【拟解决的关键问题】研究弃耕地和开垦后农田不同土层土壤细菌组成以及多样性的影响;了解某一类菌群所具有的特殊作用。分析开垦及秸秆还田对土壤微生物群落的影响,为构建健康的土壤微生态环境提供参考。
1 材料与方法
1.1 材 料
试验地点位于新疆玛纳斯河流域冲积平原十户滩镇(86°8'23"E~96°8'40"E, 44°37'29"N~44°37'49"N)。此地区位于准噶尔盆地南部,身处内陆,远离海洋,干旱少雨,蒸发量较大,年均气温6.6℃,≥10℃的积温可以达到3 490℃,年降水量110~200 mm,年蒸发量1 500~2 000 mm,无霜期148~187 d,属于典型的温带大陆性气候。
试验地选择盐渍化严重而弃耕的土地(25 hm2),弃耕了29a后进行开垦种植。
1.2 方 法
1.2.1 试验设计
试验分为2个处理,弃耕地(5 hm2)作为对照处理,植被覆盖较少,主要植被有柽柳、盐爪爪、花花柴和绢蒿等;在原始弃耕地的基础上人为开垦棉田(20 hm2)。这些处理的区域为自然的弃耕地和农田,并没有人为的控制试验地的面积。
开垦后种植棉花,种植密度为24×104株/hm2,整个作物生育期采用膜下滴灌,灌水10~12次,年灌水总量为4 500 m3/hm2。在棉花整个生育期中,纯氮(300 kg/hm2)和纯磷(200 kg/hm2)均通过膜下滴灌随水分期施用。棉花收获后,秸秆(6 000~7 500 kg/hm2)全量还田,同时尿素(150 kg/hm2)及过磷酸钙(450 kg/hm2)作为基肥深翻施入土壤。
于2016年8月棉花花期,采集各处理0~10和10~20 cm土层土样。使用土钻(0~20 cm深;直径2.5 cm)采集土样。每个处理中以“S”型,随机选取5点进行取样。将同一处理同一土层土壤样品进行充分混合,形成一个混合样品。4个土壤样品分别被装入灭菌后的自封袋中,密封后装入干冰盒中迅速带回实验室。土壤样品过2 mm筛后进行充分的混合。每一个土壤样品被分为两个部分,一部分保存于-80℃的冰箱中用于DNA的提取及后续微生物分析;另一部分自然风干用于土壤化学性质的分析。
1.2.2 土壤理化性质测定
土壤pH测定采用电极法(5∶1水/土);电导率采用电导率仪测定(2.5∶1水/土);有机质采用重铬酸钾容量法-外加热法[14];碱解氮采用碱解扩散法[15];速效磷采用0.5 mol/L NaHCO3浸提-钼锑抗比色法[16];速效钾采用NH4OAc浸提-火焰光度法[17];土壤含水量采用烘干法测定[18];微生物量碳采用氯仿熏蒸-浸提法测定[19]。
1.2.3 DNA提取、PCR扩增和Illumina MiSeq测序
根据说明书使用FastDNATMSpin Kit for Soil试剂盒(MP Biomedicals, California, USA)从0.5 g土壤样品中提取全部基因组DNA[20]。抽提后的DNA浓度使用NanoDrop 2000分光光度计进行检测。
对16S rRNA基因515-907 (V4-V5)区域进行PCR扩增[21]。上游引物通常附加一个barcode (在每个样品中含有8个独有的碱基序列): 515F (5′-barcode-GTGCCAGCMGCCGCGG-3′) and 907R (5′-CCGTCAATTCMTTTRAGTTT-3′)[21]。
PCR采用20 μL反应体系:5×FastPfu Buffer, 4 μL; 2.5 mM dNTPs, 2 μL; Forward Primer(5 μM), 0.8 μL; Reverse Primer(5 μM), 0.8 μL; FastPfu Polymerase, 0.4 μL; Template DNA, 10 ng; 补ddH2O至20 μL。PCR条件如下:1 × (95℃ 3 min); 27个循环(模板DNA变性: 95℃ 30 s; 模板DNA与引物退火: 55℃ 30 s; 引物延伸: 72℃ 45 s); 最终延伸: 72℃ 10 min(GeneAmp®9700, ABI, USA)。
使用2%琼脂糖凝胶提取扩增子,根据说明书使用AxyPrep DNA凝胶回收试剂盒(Axygen Biosciences, Union City, CA, USA)对其进行纯化,并使用QuantiFluorTM-ST蓝色荧光定量系统进行定量(Promega, USA)。将纯化的扩增子等摩尔进行混合,根据协议使用Illumina MiSeq平台对paired-end序列(2×300)进行测序(Majorbio BioPharm Technology Co., Ltd., Shanghai, China)。
1.2.4 测序数据
使用QIIME (Quantitative Insights into Microbial Ecology, version 1.9.1)对原始序列质量进行质控和过滤。遵循以下标准:(1)过滤reads尾部质量值20以下的碱基,设置50 bp的窗口,如果窗口内的平均质量值低于20,从窗口开始截去后端碱基,过滤质控后50 bp以下的reads,去除含N碱基的reads;(2)根据PE reads之间的overlap关系,将成对reads拼接(merge)成一条序列,最小overlap长度为10 bp;(3)拼接序列的overlap 区允许的最大错配比率为0.2,筛选不符合序列;(4)根据序列首尾两端的barcode和引物区分样品,并调整序列方向,barcode允许的错配数为0,最大引物错配数为2。
使用USEARCH (vsesion 7.1)在97%相似水平下对所有序列进行OTU (Operational Taxonomic Units)划分,用UCHIME检测并去除Chimeric序列[22]。为了得到每个OTU对应的物种分类信息,采用RDP Classifier (version 2.2)贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析,对比SILVA (Release 123)16S细菌核糖体数据库,置信度阈值为70%[23]。
1.3 数据处理
使用Mothur软件(version 1.30.1)对Chao1和ACE丰富度指数,Shannon和Simpson多样性指数进行计算[24]。使用Venn图描述群落共有以及独有的OTU数[25]。使用R语言进行基于Bray-Cutris距离的层次聚类分析[26]和主坐标分析(PcoA)。
使用R语言计算并作图。使用SPSS (version 19.0, SPSS Inc., Chicago, IL, USA)进行统计分析。数据分析使用单因素方差分析(one-way ANOVA),多重比较使用LSD检验(P< 0.05)。
2 结果与分析
2.1 土壤理化特性
研究表明,开垦显著降低了0~10 cm土层土壤pH,而10~20 cm变化并不显著;开垦后0~10 cm土层pH显著低于10~20 cm。开垦显著降低了各土层电导率,分别降低了80.59%和72.89%;弃耕地0~10 cm土层电导率显著高于10~20 cm,而开垦后变化趋势则相反。开垦显著增加了各土层土壤有机质、碱解氮和速效磷含量,但显著降低了速效钾含量。各土层土壤有机质、碱解氮和速效磷分别增加了243.21%和343.47%、163.15%和139.05%、123.01%和96.25%,速效钾显著降低了40.04%和38.30%。开垦前后各土层土壤有机质含量变化均不显著;开垦后0~10 cm土层碱解氮和速效磷含量均显著高于10~20 cm;开垦前后0~10 cm土层速效钾含量均显著高于10~20 cm。表1
表1 开垦前后土壤理化特性
Table 1 Soil physicochemical properties in the unfarmed and reclamation soils
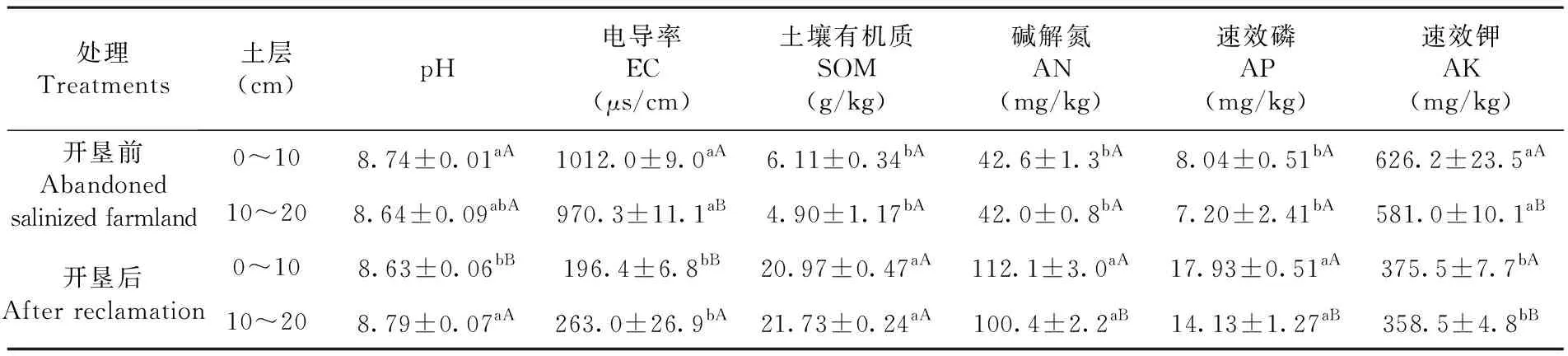
处理Treatments土层(cm)pH电导率EC(μs/cm)土壤有机质SOM(g/kg)碱解氮AN(mg/kg)速效磷AP(mg/kg)速效钾AK(mg/kg)开垦前Abandoned salinized farmland0~108.74±0.01aA1012.0±9.0aA6.11±0.34bA42.6±1.3bA8.04±0.51bA626.2±23.5aA10~208.64±0.09abA970.3±11.1aB4.90±1.17bA42.0±0.8bA7.20±2.41bA581.0±10.1aB开垦后After reclamation0~108.63±0.06bB196.4±6.8bB20.97±0.47aA112.1±3.0aA17.93±0.51aA375.5±7.7bA10~208.79±0.07aA263.0±26.9bA21.73±0.24aA100.4±2.2aB14.13±1.27aB358.5±4.8bB
注:数据为平均值±标准偏差(n=3)。不同的小写字母表示相同土层不同处理差异显著(P< 0.05),不同大写字母表示相同处理不同土层差异显著(P< 0.05)
Note: Values are means ± standard deviation (n=3). Different lowercase letters indicate significant differences among treatments within the same soil depth (P< 0.05). Different uppercase letters indicate significant differences between soil depths within the same treatments (P< 0.05)
2.2 土壤细菌α-多样性
4个样品测序共获得134 914个优化序列,包括97 000个有效序列。共划分了5 521个OTUs。每个样品的序列数范围在21 864~30 071,OTUs范围在894~1 746。与弃耕地相比,开垦后两个土层土壤样品中均观测到了更高的OTU数。弃耕地0~10 cm土层土壤样品中观测到的OTU数高于10~20 cm,而开垦后变化相反。
各土层中,开垦后Chao1和ACE指数均显著高于弃耕地。弃耕地0~10 cm土层Chao1和ACE指数均显著高于10~20 cm,而开垦后0~10 cm土层Chao1和ACE指数均显著低于10~20 cm。开垦后各土层Shannon指数显著高于弃耕地。弃耕地0~10 cm土层Shannon指数显著高于10~20 cm,而开垦后两个土层的变化趋势则相反。开垦显著降低了各土层Simpson指数。弃耕地0~10 cm土层Simpson指数显著低于10~20 cm,开垦后两个土层Simpson指数差异不显著。表2
表2 开垦前后MiSeq测序结果和多样性指数
Table 2 MiSeq sequencing results and diversity indices in the unfarmed and reclamation soils

处理Treatments土层(cm)测序结果多样性指数总序列数总OTUsChao1ACEShannonSimpson开垦前Abandoned salinized farmland0~1022 9721 2671 524±82bA1 482±54bA5.23±0.03bA0.03±0.001 2aB10~2030 0718941 070±80bB1 065±62bB3.35±0.03bB0.158 3±0.003 8aA平均值26 5221 081开垦后After reclamation0~1022 0931 6141 877±75aB1 851±53aB6.21±0.02aB0.004 9±0.0.00 02bA10~2021 8641 7462 056±79aA2 070±64aA6.29±0.03aA0.004 5±0.000 2bA平均值21 9791 680
注:数据为平均值±标准偏差(n=3)。不同的小写字母表示相同土层不同处理差异显著(P< 0.05),不同大写字母表示相同处理不同土层差异显著(P< 0.05)
Note: Values are means ± standard deviation (n=3). Different lowercase letters indicate significant differences among different treatments within the same soil depth (P< 0.05). Different uppercase letters indicate significant differences between soil depths within the same treatments (P< 0.05)
2.3 细菌类群
在0~10和10~20 cm土层中分别得到的OTU数为:1 273和894(弃耕地);1 612和1 745(开垦后)。与弃耕地相比,开垦后土壤中较多独有的OTU数(0~10 cm: 733; 10~20 cm: 1 160),开垦影响了土壤细菌类群的组成。0~10 cm土层中共有的OTU数高于10~20 cm。
弃耕地0~10 cm土层独有的OTU包括:OTU441 (TX1A-55_norank, 4.15%),OTU2312 (OM1_clade_norank, 3.84%)和OTU134 (Pseudomonas, 2.57%);10~20 cm土层独有的OTU包括:OTU304 (Bacteroides, 10.72%),OTU316 (Bacteroides, 5.85%)和OTU441 (TX1A-55_norank, 5.31%)。开垦后0~10 cm土层新产生的OTU包括:OTU2479 (Cytophagaceae_uncultured, 7.74%),OTU1629 (Anaerolineaceae_uncultured, 3.77%)和OTU1908 (Anaerolineaceae_uncultured, 2.76%);10~20 cm土层新产生的OTU包括:OTU1569 (RB41_norank, 2.78%),OTU1865 (Brevibacillus, 1.90%)和OTU2404 (Nitrospira, 1.78%)。开垦前后共有的OTU包括:OTU200 (Bacillus, 0~10 cm: 8.59%; 10~20 cm: 25.54%),OTU448 (Lactococcus, 0~10 cm: 5.75%; 10~20 cm: 15.60%)和OTU77 (Pseudomonas, 0~10 cm: 3.96%; 10~20 cm: 5.36%)。图1

注:A1和D1代表开垦前后0~10 cm土层,A2和D2代表10~20 cm土层
Note: A1 and D1 represent the 0-10 cm depth of the unfarmed and reclamation treatments, respectively. A2 and D2 represent the 10-20 cm soil depth
图1 开垦前后土壤共有和独有的OTU数
Fig.1 Number of common and unique OTUs (3% cutoff level) in the unfarmed and reclamation soils
2.4 细菌群落组成
OTU被分配到了34个不同的细菌门,研究显示了相对丰度前10的细菌门。开垦前后两个土层中Proteobacteria (22.72%~28.85%)的相对丰度最高(除了弃耕地10~20 cm土层中Firmicutes)。其它主要的细菌门包括:Firmicutes (2.96%~62.24%),Chloroflexi (2.40%~17.66%),Acidobacteria (2.32%~17.14%)和Actinobacteria (4.00%~11.70%)。开垦降低了Proteobacteria和Firmicutes的相对丰度,相反的是弃耕地Chloroflexi, Acidobacteria, Actinobacteria, Planctomycetes, Gemmatimonadetes, Bacteroidetes, Nitrospirae, and Bacteria_unclassified的相对丰度低于开垦后土壤。弃耕地0~10和10~20 cm土层Firmicutes的相对丰度变化较大。图2
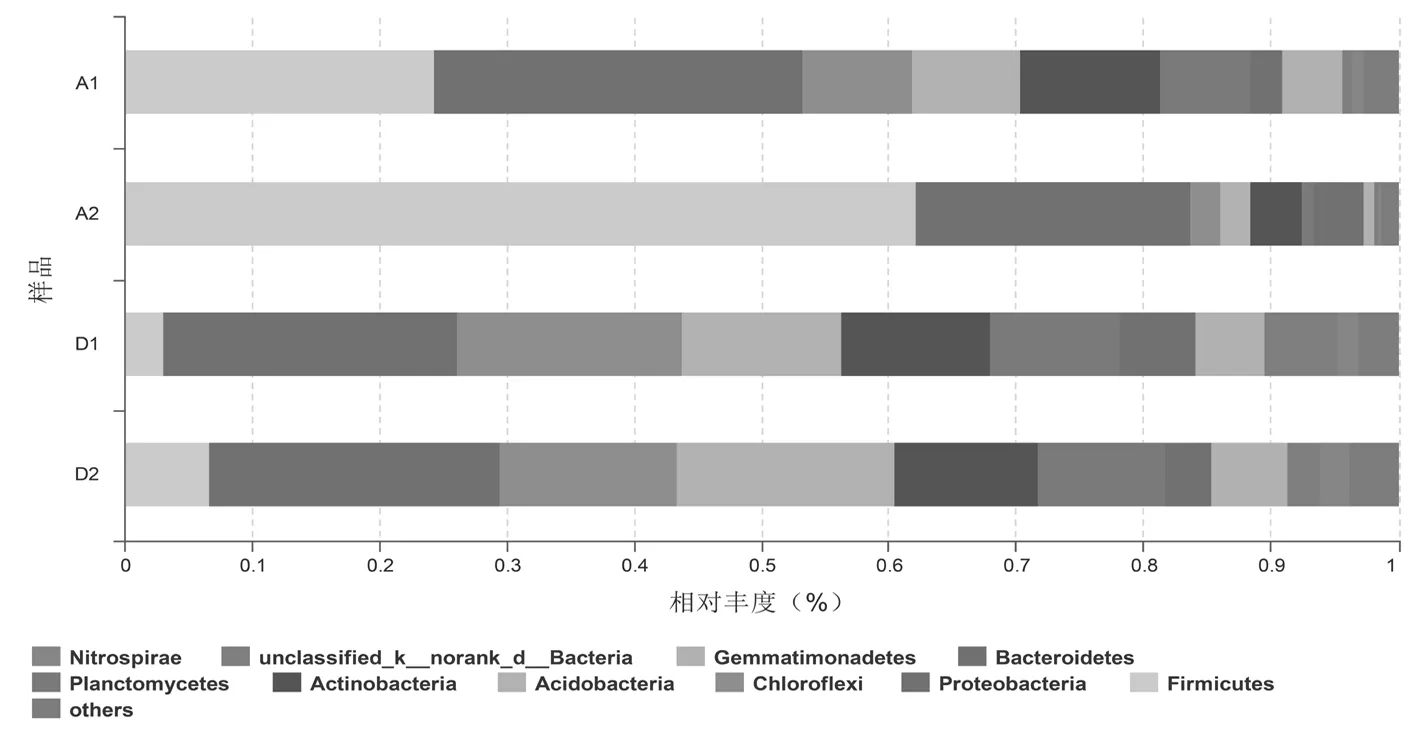
注:A1和D1代表开垦前后0~10 cm土层,A2和D2代表10~20 cm土层
Note: A1 and D1 represent the 0-10 cm depth of the unfarmed and reclamation treatments, respectively. A2 and D2 represent the 10-20 cm soil depth
图2 开垦前后相对丰度较高的10个细菌门
Fig.2 Ten bacterial phyla with the highest relative abundance in the unfarmed and reclamation soils
在属水平下,OTU被划分到506个不同的属。0~10和10~20 cm土层分别发现了341和320个属(弃耕地);380和393个属(开垦后)。优势属有:Anaerolineaceae_uncultured,Bacillus,Subgroup_6_norank,Lactococcus,Pseudomonas和Nitrosomonadaceae_uncultured。在相对丰度前10的细菌属中,开垦增加了各土层Anaerolineaceae_uncultured,Subgroup_6_norank,Nitrosomonadaceae_uncultured,Gemmatimonadaceae_uncultured,Ardenticatenia_uncultured和Bacteria_unclassified的相对丰度,相反降低了Bacillus,Lactococcus和Pseudomonas的相对丰度。只有4个细菌属的相对丰度在4个土壤样品中都大于1%,分别是:Anaerolineaceae_uncultured,Bacillus,Nitrosomonadaceae_uncultured和OM1_clade_norank。
在属水平下分析细菌的分类学组成可以为细菌门提供更为完整的信息。例如,Proteobacteria的高丰度主要是由Pseudomonas和Nitrosomonadaceae_uncultured引起的,Firmicutes的高丰度是由Bacillus和Lactococcus引起的,Chloroflexi的高丰度是由Anaerolineaceae_uncultured引起的,而Acidobacteria的高丰度是由Subgroup_6_norank造成的。图3
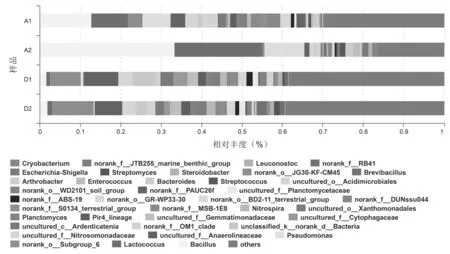
注:A1和D1代表开垦前后0~10 cm土层,A2和D2代表10~20 cm土层
Note: A1 and D1 represent the 0-10 cm depth of the unfarmed and reclamation treatments, respectively. A2 and D2 represent the 10-20 cm soil depth
图3 开垦前后相对丰度>1%细菌属
Fig.3 Bacterial genera with >1% relative abundance in the unfarmed and reclamation soils
2.5 细菌群落结构
通过对4个土壤样品OTU进行层次聚类分析。研究表明,各样品被划分为两组:(1)弃耕地;(2)开垦后农田。此外,每一个组中又包含一个处理的两个土层。PCoA是基于OTU对不同处理土壤细菌群落结构的相似性或差异性进行分析。第一和第二主成分共解释细菌群落组成92.91%的变异量。前一段中所描述的两组分别形成了一个明显的簇。主坐标轴1 (PC1)将弃耕地的细菌群落(例如:组1)与开垦后农田处理很好的分隔开来。总的来说,这些结果表明:(i)组内土壤细菌群落结构具有相似性;(ii)开垦是造成组间差异的主要原因。图4,图5
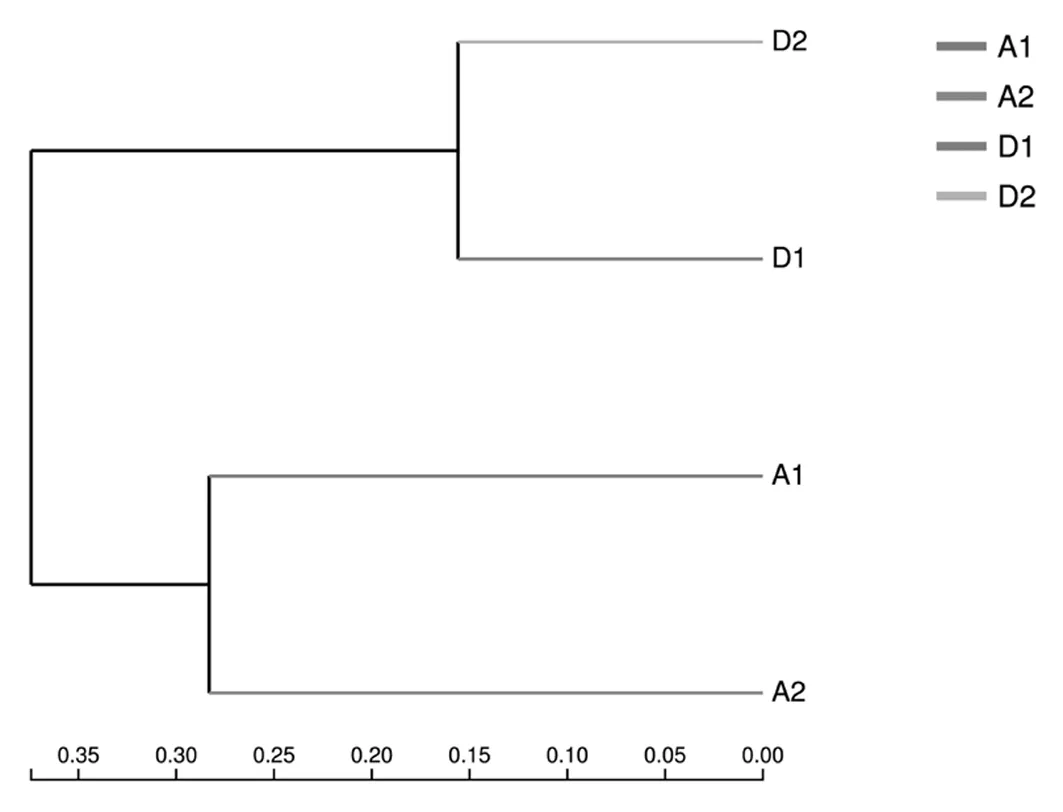
注:A1和D1代表开垦前后0~10 cm土层,A2和D2代表10~20 cm土层
Note: A1 and D1 represent the 0-10 cm depth of the unfarmed and reclamation treatments, respectively. A2 and D2 represent the 10-20 cm soil depth
图4 开垦前后OTUs层次聚类分析
Fig.4 Hierarchical cluster analysis of OTUs in the unfarmed and reclamation soils
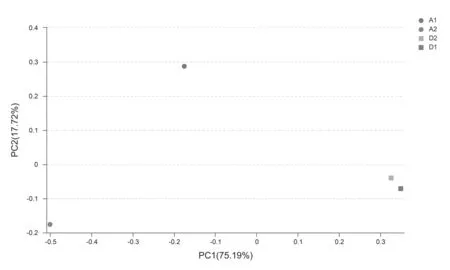
注:A1和D1代表开垦前后0~10 cm土层,A2和D2代表10~20 cm土层
Note: A1 and D1 represent the 0-10 cm depth of the unfarmed and reclamation treatments, respectively. A2 and D2 represent the 10-20 cm soil depth
图5 开垦前后OTUs主坐标分析
Fig.5 Principal coordinates analysis (PCoA) of OTUs in the unfarmed and reclamation soils
3 讨 论
3.1 土壤理化特性
与弃耕地相比,经过开垦重新种植作物后显著影响了土壤理化性质。开垦后土壤理化性质(土壤电导率、有机质、碱解氮、速效磷和速效钾)发生了显著的改变。这一系列的变化归因于每年肥料的施用以及秸秆还田,而弃耕地没有任何外源物质的供给。与弃耕地相比,开垦后显著降低了两个土层土壤电导率。在此试验区内,棉田的水分管理应用了膜下滴灌技术。“盐随水走”向下运移,并且在土壤剖面深处聚集。由于灌溉土壤盐分迅速下降,这与Zhou[27]的研究结果相一致。开垦前后两个土层中,弃耕地0~10 cm土层土壤电导率最高,说明在弃耕地随着土壤水分的蒸发,土壤盐分表现出明显的表聚现象。
开垦后土壤有机质含量显著高于弃耕地。因为研究区位于干旱地区限制了植物的生长,弃耕地植被生物量普遍偏低,土壤有机质含量较低[28]。棉花的根系有利于土壤有机质的增加,植被的凋落物是有机质的主要来源。与弃耕地相比,秸秆还田增加了土壤中的有机物质。荒地开垦后种植棉花并在秋天收获后全量秸秆还田,土壤有机质来源的一个重要途径就是植物残体。进入土壤的作物秸秆不断被分解,逐步进行矿化,秸秆中的有机物质释放到土壤中,供作物吸收利用[29],因此,秸秆还田是一种能够迅速提高土壤有机质含量的措施。棉花秸秆还田配合肥料的施用可以有效的遏制有机物质的分解,显著提高土壤有机碳[30]。开垦后土壤碱解氮和速效磷含量显著高于弃耕地。此研究结果与Zu等[31]相一致,报道称秸秆还田结合化肥的施用可以增加土壤养分含量,因此,可以很好的维持棉田生产力。在棉花整个生育期通过滴灌方式向土壤中分期施用氮肥和磷肥,这可能是造成0~10 cm土层中碱解氮和速效磷含量较高的原因。
3.2 细菌群落多样性、组成及结构
影响土壤细菌群落结构和多样性的因素有许多,包括气候、植被和土壤类型。在农业系统中,作物种类和管理措施成为影响土壤细菌群落的主要方式。序列分析显示开垦后作物种植增加了细菌物种的丰富度和多样性。研究区位于干旱地区,弃耕地没有灌溉,植被覆盖稀少。有研究称,在美国南北部pH > 8的荒漠土中细菌群落的多样性很低[32]。与弃耕地形成鲜明对比的是开垦后种植棉花引入了灌溉和施肥。碳的输入(包括根系和凋落物)相比于弃耕地有很大的提高。细菌群落对这些管理方式产生不同程度的响应,带来的变化是一些菌群增加另一些则减少。
由于开垦后进行了农业生产,使得开垦增加了土壤细菌群落的多样性。同样有研究发现开垦增加了土壤微生物多样性指数,表明荒漠的开垦有利于微生物的增长并促进其多样性的增加[33]。弃耕地开垦过程中改变了地表植被的覆盖,作物生物量、凋落物、根系等显著高于弃耕地,为土壤提供大量养分,增加了土壤碳素和氮素的供应,不仅改变了土壤微生物结构,提高了微生物生物量,还增加了土壤微生物多样性。土层深度显著地影响了土壤微生物群落丰富度和多样性指数。在半干旱地区土壤水分含量成为了不同土层深度土壤微生物群落差异的影响因素。有研究表明土壤水分的变化可以影响土壤细菌群落的组成[34]。
土壤细菌群落受多种因素的影响,土壤类型[35]、植物[36]以及耕作方式[37]等影响着土壤细菌群落结构。研究采用了“空间替代的方法”反映土壤细菌的变化与开垦之间的关系,同时减少了其它因素的干扰。聚类分析显示两个土层中弃耕地与开垦后土壤细菌群落具有差异性。Wang研究发现,荒漠开垦为农田后使土壤微生物群落结构发生显著的改变,土壤环境由贫瘠的“真菌型”向肥沃的“细菌型”转变[33]。这些研究结果表明开垦影响了土壤细菌群落结构。
Janssen通过分析各种土壤的16S rRNA基因得出土壤细菌群落主要由Proteobacteria, Acidobacteria, Actinobacteria, Verrucomicrobia, Bacteroidetes, Chloroflexi, Planctomycetes, Gemmatimonadetes和Firmicutes组成。通过序列分析得到Proteobacteria和Firmicutes是弃耕地土壤中主要的细菌门,其相对丰度占土壤细菌群落的53.11%~83.71%。同样Proteobacteria也是开垦后农田土壤主要的细菌门。开垦降低了Firmicutes的相对丰度,增加了Chloroflexi, Acidobacteria和Actinobacteria的相对丰度。这与Kob⊇rl比较了埃及沙漠土壤与其临近进行农业耕作的土壤细菌群落所得到的结果相反[38]。他们得出耕种增加了Firmicutes的相对丰度,但是降低了Proteobacteria和Actinobacteria的相对丰度。一个很重要的原因在于两个地区的土壤养分状况不同,埃及的农田富含有机物质。之所以研究者对Proteobacteria产生浓厚的兴趣有两点原因。第一,Proteobacteria可以增加植物对养分的吸收以及抵御病害的能力,从而促进作物的生长;第二,Proteobacteria涉及到甲烷和一氧化二氮两种温室气体的产生。Betaproteobacteria, Gammaproteobacteria和Firmicutes可以抑制土传病害的发生[39]。
4 结 论
对于本底养分低的荒地,开垦显著降低了土壤电导率,降低了72.89%~80.59%。开垦显著增加了各土层土壤有机质、碱解氮和速效磷含量,但显著降低了速效钾含量。各土层土壤有机质、碱解氮和速效磷分别增加了243.21%和343.47%、163.15%和139.05%、123.01%和96.25%,速效钾降低了40.04%和38.30%。盐碱弃耕地开垦为棉田后显著的增加了土壤细菌群落的丰富度以及多样性,改变了菌群的结构。Proteobacteria和Firmicutes是弃耕地土壤中主要的细菌门。开垦降低了Firmicutes的相对丰度,但是增加了Chloroflexi, Acidobacteria和Actinobacteria的相对丰度。弃耕地的开垦有利于微生物的生长,促进其多样性。
参考文献(References)
[1] Zhao, J., Zhang, R., Xue, C., Xun, W., Sun, L., & Xu, Y., et al. (2014). Pyrosequencing reveals contrasting soil bacterial diversity and community structure of two main winter wheat cropping systems in china.MicrobialEcology, 67(2): 443-453.
[2] Zhong, W., Gu, T., Wang, W., Zhang, B., Lin, X., & Huang, Q., et al. (2010). The effects of mineral fertilizer and organic manure on soil microbial community and diversity.Plant&Soil, 326(1-2): 511-523.
[3] Paul, E. A. (2014). Soil microbiology and biochemistry.JournalofRangeManagement, 51(2): 254.
[4] Quadros, P. D. D., Zhalnina, K., Davisrichardson, A., Fagen, J. R., Drew, J., & Bayer, C., et al. (2012). The effect of tillage system and crop rotation on soil microbial diversity and composition in a subtropical acrisol.Diversity, 4(4): 375-395.
[5] Qiu, M., Zhang, R., Xue, C., Zhang, S., Li, S., & Zhang, N., et al. (2012). Application of bio-organic fertilizer can control fusarium, wilt of cucumber plants by regulating microbial community of rhizosphere soil.Biology&FertilityofSoils, 48(7): 807-816.
[6] Johnsen, K., Jacobsen, C. S., Torsvik, V., & Sorenson, A. J. (2001). Pesticide effects of bacterial diversity in agricultural soils--a review.Biology&FertilityofSoils, 33(6): 443-453.
[7] Rietz, D. N., & Haynes, R. J. (2003). Effects of irrigation-induced salinity and sodicity on soil microbial activity.SoilBiology&Biochemistry, 35(6): 845-854.
[8] Liu, E., Yan, C., Mei, X., He, W., Bing, S. H., & Ding, L., et al. (2010). Long-term effect of chemical fertilizer, straw, and manure on soil chemical and biological properties in northwest china.Geoderma, 158(3-4): 173-180.
[9] Cui, J., Liu, C., Li, Z., Wang, L., Chen, X., & Ye, Z., et al. (2012). Long-term changes in topsoil chemical properties under centuries of cultivation after reclamation of coastal wetlands in the yangtze estuary, china.Soil&TillageResearch, 123(5): 50-60.
[10] Hartmann, M., Lee, S., Hallam, S., & Mohn, W. (2009). Bacterial, archaeal and eukaryal community structures throughout soil horizons of harvested and naturally disturbed forest stands.EnvironmentalMicrobiology, 11(12): 3,045-3,062.
[11] Will, C., Thürmer, A., Wollherr, A., Nacke, H., Herold, N., & Schrumpf, M., et al. (2010). Horizon-specific bacterial community composition of german grassland soils, as revealed by pyrosequencing-based analysis of 16s rrna genes.Applied&EnvironmentalMicrobiology, 76(20): 6,751-6,759.
[12] Oberauner, L., Zachow, C., Lackner, S., H?genauer, C., Smolle, K. H., & Berg, G. (2013). The ignored diversity: complex bacterial communities in intensive care units revealed by 16s pyrosequencing.SciRep, 3(3):1,413.
[13] Sun, Y., Cai, Y., Liu, L., Yu, F., & Farmerie, W. (2009). Esprit: estimating species richness using large collections of 16s rrna data.NucleicAcidsResearch, 37(10):e76.
[14] ClaudioCiavatta, MarcoGovi, LiviaVittoriAntisari, & PaoloSequi. (1991). Determination of organic carbon in aqueous extracts of soils and fertilizers.CommunicationsinSoilScience&PlantAnalysis, 22(9-10):795-807.
[15] Mulvaney, R. L., & Khan, S. A. (2001). Diffusion methods to determine different forms of nitrogen in soil hydrolysates.SoilScienceSocietyofAmericaJournal, 65(4): 1,284-1,292.
[16] Page, A. L., Miller, R. H., & Keeney, D. R. (1982).Chemicalandmicrobiologicalproperties. American Society of Agronomy : Soil Science Society of America.
[17] Shen, Z., Zhong, S., Wang, Y., Wang, B., Mei, X., & Li, R., et al. (2013). Induced soil microbial suppression of banana fusarium wilt disease using compost and biofertilizers to improve yield and quality.EuropeanJournalofSoilBiology, 57(4):1-8.
[18] Yang, W., Zhao, H., Chen, X., Yin, S., Cheng, X., & An, S. (2013). Consequences of short-term c 4, plant spartina alterniflora, invasions for soil organic carbon dynamics in a coastal wetland of eastern china.EcologicalEngineering, 61(12): 50-57.
[19] Vance, E. D., Brooks, P. C., & Jenkinson, D. S. (1987). An extraction method for measuring soil microbial biomass.SoilBiology&Biochemistry, 19(19): 703-707.
[20] Zhou, J., Bruns, M. A., & Tiedje, J. M. (1996). Dna recovery from soils of diverse composition.ApplEnvironMicrobiol, 62(2): 316-322.
[21] Xiong, J., Liu, Y., Lin, X., Zhang, H., Zeng, J., & Hou, J., et al. (2012). Geographic distance and ph drive bacterial distribution in alkaline lake sediments across tibetan plateau.EnvironmentalMicrobiology, 14(9):2,457-2,466.
[22] Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., & Knight, R. (2011). Uchime improves sensitivity and speed of chimera detection.Bioinformatics, 27(16): 2,194-2,200.
[23] Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., & Yarza, P., et al. (2013). The silva ribosomal rna gene database project: improved data processing and web-based tools.NucleicAcidsResearch,41(Database issue): 590-596.
[24] Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., & Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities.Applied&EnvironmentalMicrobiology, 75(23): 7,537-7,541.
[25] Fouts, D. E., Szpakowski, S., Purushe, J., Torralba, M., Waterman, R. C., & Macneil, M. D., et al. (2012). Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen.PlosOne, 7(11): e48289.
[26] Jiang, X. T., Peng, X., Deng, G. H., Sheng, H. F., Wang, Y., & Zhou, H. W., et al. (2013). Illumina sequencing of 16s RRNA tag revealed spatial variations of bacterial communities in a mangrove wetland.MicrobialEcology, 66(1): 96-104.
[27] 周丽, 王玉刚, 李彦,等. 盐碱荒地开垦年限对表层土壤盐分的影响[J]. 干旱区地理, 2013, 36(2): 285-291.
ZHOU Li, WANG Yu-gang, LI Yan, et al. (2013). Effects of cultivation on soil salinity in upper soil profiles of the saline-alkali land[J].AridLandGeography, 36(2): 285-291. (in Chinese)
[28] Zhang, F. H., Yang, H. C., Gale, W. J., Cheng, Z. B., & Yan, J. H. (2016). Temporal changes in soil organic carbon and aggregate-associated organic carbon after reclamation of abandoned, salinized farmland.JournalofAgriculturalScience, 155(2): 205-215.
[29] 王双磊. 棉花秸秆还田对盐碱地棉田土壤理化性质和生物学特性的影响[D]. 泰安:山东农业大学硕士学位论文, 2015.
WANG Shuang-lei. (2015).Effectofcottonstrawincorporatedintosaline-alkalisoilonphysical,chemicalandbiologicalproperties[D]. Master Dissertation. Shandong Agricultural University, Taian. (in Chinese)
[30] 姜益娟, 郑德明, 吕双庆, 等. 连续施用棉籽饼和棉秆还田及化肥配施的培肥效应[J]. 干旱地区农业研究, 1999,(4): 19-24.
JIANG Yi-juan, ZHENG De-ming, LÜ Shuang-qin, ,et al. (1999). Fertilization effect of continuous application of cotton seed manure and cotton stalk returned to field together with combined application of chemical fertilizers [J].AgriculturalResearchinTheAridAreas, (4): 19-24. (in Chinese)
[31] Zu, C., Li, Z. G., Yang, J. F., Yu, H., Sun, Y., & Tang, H. L., et al. (2014). Acid soil is associated with reduced yield, root growth and nutrient uptake in black pepper (piper nigrum l.).JournalofAgriculturalScience, 5(5): 466-473.
[32] Lauber, C. L., Hamady, M., Knight, R., & Fierer, N. (2009). Pyrosequencing-based assessment of soil ph as a predictor of soil bacterial community structure at the continental scale.Applied&EnvironmentalMicrobiology, 75(15): 5,111-5,120.
[33] 王银亚, 李晨华, 马健. 开垦对荒漠土壤微生物群落结构特征的影响[J]. 中国沙漠, 2017, 37(3): 514-522.
WANG Yin-ya, LI Chen-hua, MA Jian. (2017). Effects of Desert Rclamation on Soil Microbial ommunity and Microbial Diversity [J].JournalofDesertResearch, 37(3): 514-522. (in Chinese)
[34] Wilkinson, S. C., Anderson, J. M., Scardelis, S. P., Tisiafouli, M., Taylor, A., & Wolters, V. (2002). Plfa profiles of microbial communities in decomposing conifer litters subject to moisture stress.SoilBiology&Biochemistry, 34(2): 189-200.
[35] Garbeva, P., van Veen, J. A., & van Elsas, J. D. (2004). Microbail diversity in soil: selection of microbial populations by plant and soil type and implications for disease suppressiveness.AnnualReviewofPhytopathology, 42(42): 243-270.
[36] Wieland, G., Neumann, R., & Backhaus, H. (2001). Variation of microbial communities in soil, rhizosphere, and rhizoplane in response to crop species, soil type, and crop development.Applied&EnvironmentalMicrobiology, 67(12):5,849-5,854.
[37] Sudini, H., Liles, M. R., Arias, C. R., Bowen, K. L., & Huettel, R. N. (2011). Exploring soil bacterial communities in different peanut-cropping sequences using multiple molecular approaches.Phytopathology, 101(7): 819-827.
[38] Kberl, M., Müller, H., Ramadan, E. M., & Berg, G. (2011). Desert farming benefits from microbial potential in arid soils and promotes diversity and plant health.PlosOne, 6(9): e24452.
[39] Mendes, R., Kruijt, M., De, B. I., Dekkers, E., Van, d. V. M., & Schneider, J. H., et al. (2011). Deciphering the rhizosphere microbiome for disease-suppressive bacteria.Science, 332(6033):1,097-1,100.
